
Chloride channels are a superfamily of poorly understood ion channels specific for chloride. These channels may conduct many different ions, but are named for chloride because its concentration in vivo is much higher than other anions. Several families of voltage-gated channels and ligand-gated channels have been characterized in humans.
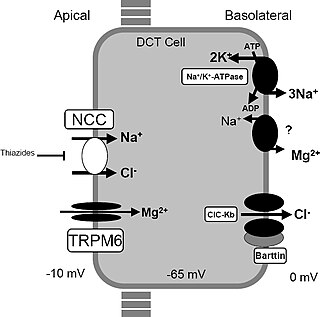
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH. It is the most frequent hereditary salt-losing tubulopathy. Gitelman syndrome is caused by disease-causing variants on both alleles of the SLC12A3 gene. The SLC12A3 gene encodes the thiazide-sensitive sodium-chloride cotransporter, which can be found in the distal convoluted tubule of the kidney.
The renal outer medullary potassium channel (ROMK) is an ATP-dependent potassium channel (Kir1.1) that transports potassium out of cells. It plays an important role in potassium recycling in the thick ascending limb (TAL) and potassium secretion in the cortical collecting duct (CCD) of the nephron. In humans, ROMK is encoded by the KCNJ1 gene. Multiple transcript variants encoding different isoforms have been found for this gene.

Fibroblast growth factor 23 (FGF23) is a protein and member of the fibroblast growth factor (FGF) family which participates in the regulation of phosphate in plasma and vitamin D metabolism. In humans it is encoded by the FGF23 gene. FGF23 decreases reabsorption of phosphate in the kidney. Mutations in FGF23 can lead to its increased activity, resulting in autosomal dominant hypophosphatemic rickets.
The sodium-chloride symporter (also known as Na+-Cl− cotransporter, NCC or NCCT, or as the thiazide-sensitive Na+-Cl− cotransporter or TSC) is a cotransporter in the kidney which has the function of reabsorbing sodium and chloride ions from the tubular fluid into the cells of the distal convoluted tubule of the nephron. It is a member of the SLC12 cotransporter family of electroneutral cation-coupled chloride cotransporters. In humans, it is encoded by the SLC12A3 gene (solute carrier family 12 member 3) located in 16q13.

Dent's disease is a rare X-linked recessive inherited condition that affects the proximal renal tubules of the kidney. It is one cause of Fanconi syndrome, and is characterized by tubular proteinuria, excess calcium in the urine, formation of calcium kidney stones, nephrocalcinosis, and chronic kidney failure.
Transient receptor potential cation channel subfamily V member 5 is a calcium channel protein that in humans is encoded by the TRPV5 gene.
The CLCN family of voltage-dependent chloride channel genes comprises nine members which demonstrate quite diverse functional characteristics while sharing significant sequence homology. The protein encoded by this gene regulates the electric excitability of the skeletal muscle membrane. Mutations in this gene cause two forms of inherited human muscle disorders: recessive generalized myotonia congenita (Becker) and dominant myotonia (Thomsen).

The CLCN5 gene encodes the chloride channel Cl-/H+ exchanger ClC-5. ClC-5 is mainly expressed in the kidney, in particular in proximal tubules where it participates to the uptake of albumin and low-molecular-weight proteins, which is one of the principal physiological role of proximal tubular cells. Mutations in the CLCN5 gene cause an X-linked recessive nephropathy named Dent disease characterized by excessive urinary loss of low-molecular-weight proteins and of calcium (hypercalciuria), nephrocalcinosis and nephrolithiasis.

Chloride channel protein 2 is a protein that in humans is encoded by the CLCN2 gene. Mutations of this gene have been found to cause leukoencephalopathy and Idiopathic generalised epilepsy, although the latter claim has been disputed. CLCN2 contains a transmembrane region that is involved in chloride ion transport as well two intracellular copies of the CBS domain.

Chloride channel Kb, also known as CLCNKB, is a protein which in humans is encoded by the CLCNKB gene.

Anion exchange protein 3 is a membrane transport protein that in humans is encoded by the SLC4A3 gene. AE3 is functionally similar to the Band 3 Cl−/HCO3− exchange protein but it is expressed primarily in brain neurons and in the heart. Like AE2 its activity is sensitive to pH. AE3 mutations have been linked to seizures.
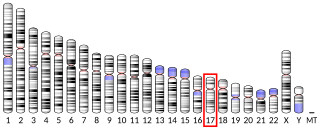
Serine/threonine protein kinase WNK4 also known as WNK lysine deficient protein kinase 4 or WNK4, is an enzyme that in humans is encoded by the WNK4 gene. Missense mutations cause a genetic form of pseudohypoaldosteronism type 2, also called Gordon syndrome.

Chloride intracellular channel protein 2 is a protein that in humans is encoded by the CLIC2 gene.
Chloride transport protein 6 is a protein that in humans is encoded by the CLCN6 gene.
Solute carrier family 22, member 12, also known as SLC22A12 and URAT1, is a protein which in humans is encoded by the SLC22A12 gene.
The Cl−-formate exchanger, otherwise known as Pendrin encoded by the SLC26A4 gene, is a transport protein present in the kidney, where it functions in the renal chloride reabsorption. It is also present in vascular smooth muscle and cardiac muscle.
Bartter syndrome, infantile, with sensorineural deafness (Barttin), also known as BSND, is a human gene which is associated with Bartter syndrome.

Solute carrier family 12 member 6 is a protein that in humans is encoded by the SLC12A6 gene.

Chloride intracellular channel protein 5 is a protein that in humans is encoded by the CLIC5 gene.




