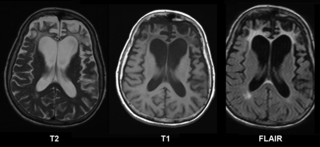
Frontotemporal dementia (FTD), also called frontotemporal degeneration disease or frontotemporal neurocognitive disorder, encompasses several types of dementia involving the progressive degeneration of the brain's frontal and temporal lobes. Men and women appear to be equally affected. FTD generally presents as a behavioral or language disorder with gradual onset. Signs and symptoms tend to appear in late adulthood, typically between the ages of 45 and 65, although it can affect people younger or older than this. Currently, no cure or approved symptomatic treatment for FTD exists, although some off-label drugs and behavioral methods are prescribed.

Amyloid beta denotes peptides of 36–43 amino acids that are the main component of the amyloid plaques found in the brains of people with Alzheimer's disease. The peptides derive from the amyloid-beta precursor protein (APP), which is cleaved by beta secretase and gamma secretase to yield Aβ in a cholesterol-dependent process and substrate presentation. Both neurons and oligodendrocytes produce and release Aβ in the brain, contributing to formation of amyloid plaques. Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms. It is now believed that certain misfolded oligomers can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prion infection. The oligomers are toxic to nerve cells. The other protein implicated in Alzheimer's disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.
In cell biology, microtubule-associated proteins (MAPs) are proteins that interact with the microtubules of the cellular cytoskeleton. MAPs are integral to the stability of the cell and its internal structures and the transport of components within the cell.

Amyloid plaques are extracellular deposits of the amyloid beta (Aβ) protein mainly in the grey matter of the brain. Degenerative neuronal elements and an abundance of microglia and astrocytes can be associated with amyloid plaques. Some plaques occur in the brain as a result of aging, but large numbers of plaques and neurofibrillary tangles are characteristic features of Alzheimer's disease. The plaques are highly variable in shape and size; in tissue sections immunostained for Aβ, they comprise a log-normal size distribution curve, with an average plaque area of 400-450 square micrometers (μm2). The smallest plaques, which often consist of diffuse deposits of Aβ, are particularly numerous. Plaques form when Aβ misfolds and aggregates into oligomers and longer polymers, the latter of which are characteristic of amyloid.

Frontotemporal lobar degeneration (FTLD) is a pathological process that occurs in frontotemporal dementia. It is characterized by atrophy in the frontal lobe and temporal lobe of the brain, with sparing of the parietal and occipital lobes.

Neurofibrillary tangles (NFTs) are intracellular aggregates of hyperphosphorylated tau protein that are most commonly known as a primary biomarker of Alzheimer's disease. Their presence is also found in numerous other diseases known as tauopathies. Little is known about their exact relationship to the different pathologies.

Corticobasal degeneration (CBD) is a rare neurodegenerative disease involving the cerebral cortex and the basal ganglia. CBD symptoms typically begin in people from 50 to 70 years of age, and typical survival before death is eight years. It is characterized by marked disorders in movement and cognition, and is classified as one of the Parkinson plus syndromes. Diagnosis is difficult, as symptoms are often similar to those of other disorders, such as Parkinson's disease, progressive supranuclear palsy, and dementia with Lewy bodies, and a definitive diagnosis of CBD can only be made upon neuropathologic examination.
The major prion protein (PrP) is encoded in the human body by the PRNP gene also known as CD230. Expression of the protein is most predominant in the nervous system but occurs in many other tissues throughout the body.

Tauopathies are a class of neurodegenerative diseases characterized by the aggregation of abnormal tau protein. Hyperphosphorylation of tau proteins causes them to dissociate from microtubules and form insoluble aggregates called neurofibrillary tangles. Various neuropathologic phenotypes have been described based on the anatomical regions and cell types involved as well as the unique tau isoforms making up these deposits. The designation 'primary tauopathy' is assigned to disorders where the predominant feature is the deposition of tau protein. Alternatively, diseases exhibiting tau pathologies attributed to different and varied underlying causes are termed 'secondary tauopathies'. Some neuropathologic phenotypes involving tau protein are Alzheimer's disease, frontotemporal dementia, progressive supranuclear palsy, and corticobasal degeneration.

A neurodegenerative disease is caused by the progressive loss of neurons, in the process known as neurodegeneration. Neuronal damage may also ultimately result in their death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic.Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) is an autosomal dominant neurodegenerative tauopathy and Parkinson plus syndrome. FTDP-17 is caused by mutations in the MAPT gene located on the q arm of chromosome 17, and has three cardinal features: behavioral and personality changes, cognitive impairment, and motor symptoms. FTDP-17 was defined during the International Consensus Conference in Ann Arbor, Michigan, in 1996.
The biochemistry of Alzheimer's disease, the most common cause of dementia, is not yet very well understood. Alzheimer's disease (AD) has been identified as a proteopathy: a protein misfolding disease due to the accumulation of abnormally folded amyloid beta (Aβ) protein in the brain. Amyloid beta is a short peptide that is an abnormal proteolytic byproduct of the transmembrane protein amyloid-beta precursor protein (APP), whose function is unclear but thought to be involved in neuronal development. The presenilins are components of proteolytic complex involved in APP processing and degradation.

In medicine, proteinopathy, or proteopathy, protein conformational disorder, or protein misfolding disease, is a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body.

Microtubule-associated protein 4 is a protein that in humans is encoded by the MAP4 gene.

Serine/threonine-protein kinase MARK1 is an enzyme that in humans is encoded by the MARK1 gene.

Phosphorylase b kinase gamma catalytic chain, skeletal muscle isoform is an enzyme that in humans is encoded by the PHKG1 gene.
Transmembrane protein 106B is a protein that is encoded by the TMEM106B gene. It is found primarily within neurons and oligodendrocytes in the central nervous system with its subcellular location being in lysosomal membranes. TMEM106B helps facilitate important functions for maintaining a healthy lysosome, and therefore certain mutations and polymorphisms can lead to issues with proper lysosomal function. Lysosomes are in charge of clearing out mis-folded proteins and other debris, and thus, play an important role in neurodegenerative diseases that are driven by the accumulation of various mis-folded proteins and aggregates. Due to its impact on lysosomal function, TMEM106B has been investigated and found to be associated to multiple neurodegenerative diseases.
Maria Grazia Spillantini, is Professor of Molecular Neurology in the Department of Clinical Neurosciences at the University of Cambridge. She is most noted for identifying the protein alpha-synuclein as the major component of Lewy bodies, the characteristic protein deposit found in the brain in Parkinson's disease and dementia with Lewy bodies. She has also identified mutations in the MAPT gene as a heritable cause for frontotemporal dementia.
Primary age-related tauopathy (PART) is a neuropathological designation introduced in 2014 to describe the neurofibrillary tangles (NFT) that are commonly observed in the brains of normally aged and cognitively impaired individuals that can occur independently of the amyloid plaques of Alzheimer's disease (AD). The term and diagnostic criteria for PART were developed by a large group of neuropathologists, spearheaded by Drs. John F. Crary and Peter T. Nelson. Despite some controversy, the term PART has been widely adopted, with the consensus criteria cited over 1130 times as of April 2023 according to Google Scholar.

Neurotubules are microtubules found in neurons in nervous tissues. Along with neurofilaments and microfilaments, they form the cytoskeleton of neurons. Neurotubules are undivided hollow cylinders that are made up of tubulin protein polymers and arrays parallel to the plasma membrane in neurons. Neurotubules have an outer diameter of about 23 nm and an inner diameter, also known as the central core, of about 12 nm. The wall of a neurotubule is about 5 nm in width. There is a non-opaque clear zone surrounding the neurotubule and it is about 40 nm in diameter. Like microtubules, neurotubules are greatly dynamic and their length can be adjusted by polymerization and depolymerization of tubulin.







