Gene and protein structure
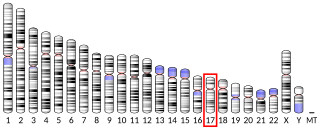
MYH9 is a large gene spanning more than 106 kilo base pairs on chromosome 22q12.3. It is composed of 41 exons with the first ATG of the open reading frame localized in exon 2 and the stop codon in exon 41. It encodes non-muscle myosin heavy chain IIA (NMHC IIA), a protein of 1,960 amino acids. Consistent with its wide expression in cells and tissues, the promoter region of MYH9 is typical of housekeeping genes having no TATA box but high GC content, with multiple GC boxes. MYH9 is a well-conserved gene through evolution. The mouse ortholog (Myh9) is localized in a syntenic region on chromosome 15 and has the same genomic organization as that of the human gene. It encodes a protein of the same length, with 97.1% amino acid identity with the human MYH9 protein. [15]
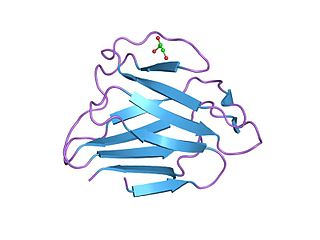
Like all class II myosins, the two NMHC IIAs dimerize producing an asymmetric molecular structure recognizable by two heads and a tail domain: the N-terminal half of each heavy chain generates the head domain, which consists of the globular motor domain and the neck domain, and the C-terminal halves of the two heavy chains together form the tail domain. [16] The motor domain, which is organized into four subdomains (SH3-like motif, the upper and the lower 50kDa subdomains, and the converter region) connected by flexible linkers, [17] interacts with filamentous actin to generate force through magnesium-dependent hydrolysis of ATP. The neck acts as a lever arm that amplifies the movement produced by conformational changes of the motor domain, and is the binding site for the light chains through two IQ motifs. The tail domain is fundamental for both dimerization of the heavy chains and formation of NM IIA functional filaments. Two heavy chains dimerize through the tail domain forming a long alpha-helical coiled-coil rod composed of typical heptad repeats. Dimers self-associate though the coiled-coil rods to form myosin filaments. The tail domain ends at the C-terminus with a 34-residue non-helical tailpiece. [14] [16]
Regulation of NM IIA structure and function
There are three paralogs of non-muscle myosin II (NM II), NM IIA, IIB, and IIC, with each having the heavy chain encoded on a different chromosome. All three paralogs appear to bind the same or very similar light chains and share basic properties as to structure and activation, but all three play distinct roles during vertebrate development and adulthood (for general reviews on NM IIs, see [11] [13] [14] ). All NM IIs have two important features: they are MgATPase enzymes that can hydrolyze ATP thereby converting chemical energy into mechanical movement. In addition, they can form bipolar filaments which can interact with and exert tension on actin filaments. These properties provide the basis for all NM II functions. The path to myosin filament formation, which is shared by NM II and smooth muscle myosin, starts with a folded inactive conformation of the NM II monomer which, upon phosphorylation of the 20 KDa light chain unfolds the molecule to produce a globular head region followed by an extended alpha-helical coiled-coil tail. [18] [19] [20] [21] The tail portion of the molecule can interact with other NM IIA hexamers to form bipolar filaments composed of 14-16 molecules.
Phosphorylation of the 20 KDa light chains on Serine 19 and Threonine 18 by a number of different kinases, but most prominently by Rho-dependent kinase and/or by the calcium-calmodulin-dependent myosin light chain kinase, not only linearizes the folded structure but removes the inhibition imposed on the MgATPase activity due to the folded conformation. In addition to phosphorylation of the 20 KDa light chains, the NMHC IIs can also be phosphorylated, but the sites phosphorylated differ among the paralogs. [10] In most cases phosphorylation of NMHC IIA can act to either dissociate the myosin filaments or to prevent filament formation.
In addition to phosphorylation, NM IIA filament assembly and localization can be modulated by interaction with other proteins including S100A4 and Lethal giant larvae (Lgl1). The former is a calcium binding protein and is also known as metastatin, a well-characterized metastatic factor. S100A4 expression is associated with enhanced cell migration through maintenance of cell polarization and inhibition of cell turning. [22] [23] Similar to heavy chain phosphorylation, in vitro binding of S100A to the carboxy-terminal end of the NM IIA coiled-coil region prevents filament formation and S100A4 binding to previously formed filaments promotes filament disassembly. The tumor suppressor protein Lgl1 also inhibits the ability of NM IIA to assemble into filaments in vitro. [24] [25] In addition, it regulates the cellular localization of NM IIA and contributes to the maturation of focal adhesions. Other proteins that are known to interact with NM IIA include the actin binding protein tropomyosin 4.2 [26] and a novel actin stress fiber associated protein, LIM and calponin-homology domains1 (LIMCH1). [27]
Clinical significance
MYH9-related disease. Mutations in MYH9 cause a Mendelian autosomal-dominant disorder known as MYH9-related disease (MYH9-RD). [36] [37] [38] [39] All affected individuals present congenital hematological alterations consisting in thrombocytopenia, platelet macrocytosis, and inclusions of the MYH9 protein in the cytoplasm of granulocytes. Most patients develop one or more non-congenital manifestations, including sensorineural deafness, kidney damage, presenile cataracts, and/or elevation of liver enzymes. [39] [40] [41] The term MYH9-RD encompasses four syndromic pictures that were considered for many years as distinct disorders, namely May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome. After the identification of MYH9 as the gene responsible for all of these entities, it was recognized that they actually represent different clinical presentations of the same disease, now known as MYH9-RD or MYH9 disorder. [38] MYH9-RD is a rare disease: prevalence is estimated around 3:1,000,000. The actual prevalence is expected to be higher, as mild forms are often discovered incidentally and patients are frequently misdiagnosed with other disorders. The disease has been reported worldwide and there is no evidence of variation in prevalence across ethnic populations. [42]
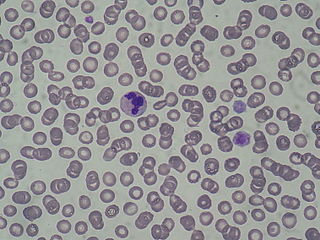
Thrombocytopenia can result in a variable degree of bleeding tendency. The majority of patients have no spontaneous bleeding or only mild cutaneous bleeding (easy bruising) and are at risk of significant hemorrhages only after surgery or other invasive procedures, deliveries, or trauma. Some patients have spontaneous mucosal bleeding, such as menorrhagia, epistaxis, and gum bleeding. [39] [40] Severe and life-threatening hemorrhages are not frequent. Platelets of MYH9-RD patients are characterized by a very large size: platelets larger than red blood cells (called "giant platelets") are always present at the examination of peripheral blood smears. [38] [43] Granulocyte inclusions of the NMHC IIA may be evident at the analysis of blood films after conventional staining as cytoplasmic basophilic (light blue) inclusions, called "Döhle-like bodies". [38] [39] More than 50% of MYH9-RD patients develop sensorineural hearing loss. [40] Severity of the hearing impairment is greatly variable, as it ranges from a mild hearing defect that occurs in mid or advanced age to a progressive hearing loss that is evident in the first years of life and rapidly evolves to severe deafness. [44] Kidney damage occurs in about 25% of patients. It presents with proteinuria and often progresses to kidney failure, which, in its most severe forms, may require dialysis and/or kidney transplantation. [40] Around 20% of patients develop presenile cataracts. About 50% of MYH9-RD patients present chronic or intermittent elevation of liver transaminases or gamma-glutamyl transferases: this alteration appears to be benign, as no patients showed evolution to liver dysfunction. [41]
Diagnosis of MYH9-RD is confirmed by the identification of the NMHC IIA inclusions in granulocytes through an immunofluorescence assay on peripheral blood smears and/or by the detection of the causative mutation through mutational screening of the MYH9 gene. [45] [46] [47] [48]
In most cases, MYH9-RD is caused by missense mutations affecting the head or tail domain of the NMHC IIA. Nonsense or frameshift alterations resulting in the deletion of a C-terminal fragment of the NMHC IIA (17 to 40 residues) are involved in approximately 20% of families. In-frame deletions or duplications have been identified in a few cases. [40] [45] [49] The disease is transmitted as an autosomal-dominant trait, however, about 35% of index cases are sporadic. [46] Sporadic forms mainly derive from de novo mutations; rare cases have been explained by germinal or somatic mosaicism. [50] [51] [52]
The incidence and the severity of the non-congenital manifestations of MYH9-RD correlate with the specific MYH9 mutation. The recent definition of genotype-phenotype correlations allows prediction of the clinical evolution of the disease in most cases. [40] [53] Genotype-phenotype correlations have been reported also for the severity of thrombocytopenia, platelet size, and the features of leukocyte inclusions. [40] [43] [54]
Within a phase 2 trial, eltrombopag, an agonist of the thrombopoietin receptor, significantly increased platelet count in 11 out of 12 patients affected with MYH9-RD. [55] ACE-inhibitors or angiotensin II receptor blockers may be effective in reducing proteinuria when given at the early stage of kidney involvement. [56] [57] Cochlear implantation is effective in restoring hearing function in MYH9-RD patients with severe/profound deafness. [58]
Role of MYH9 variants in other human diseases. Evidence obtained in animals indicates that MYH9 acts as a tumor suppressor gene. Silencing of Myh9 in the epithelial cells in mice was associated with the development of squamous cell carcinoma (SCC) of the skin and the head and neck. [59] In another mouse model, ablation of Myh9 in the tongue epithelium led to the development of tongue SCC. [60] In mice predisposed to invasive lobular breast carcinoma (ILBC) because of E-cadherin ablation, the inactivation of Myh9 led to the development of tumors recapitulating the features of human ILBC. [61] Some observations suggest that defective MYH9 expression is associated with oncogenesis and/or tumor progression in human SCC and ILBC, thus also supporting a role for MYH9 as a tumor suppressor in humans. [59] [61]
Genetic variations in MYH9 may be involved in predisposition to chronic kidney disease (CKD). A haplotype of MYH9 (haplotype E1) was previously associated with the increased prevalence of glomerulosclerosis [62] and non-diabetic end stage renal disease [63] in African Americans and in Hispanic Americans. [64] However, subsequent studies showed that this association is explained by strong linkage disequilibrium with two haplotypes (haplotypes G1 and G2) in the neighboring APOL1 gene. [65] [66] [67] Nevertheless, some studies suggest an association of single-nucleotide polymorphisms in MYH9 with CKD that appears to be independent of the linkage with APOL1 G1 and G2. [68] [69] [70]
Inherited MYH9 mutations may be responsible for non-syndromic hearing loss. [71] [72] [73]