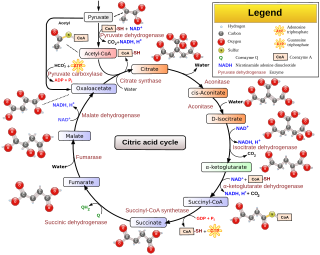
The citric acid cycle—also known as the Krebs cycle, Szent–Györgyi–Krebs cycle, or TCA cycle —is a series of biochemical reactions to release the energy stored in nutrients through the oxidation of acetyl-CoA derived from carbohydrates, fats, proteins, and alcohol. The chemical energy released is available in the form of ATP. The Krebs cycle is used by organisms that respire to generate energy, either by anaerobic respiration or aerobic respiration. In addition, the cycle provides precursors of certain amino acids, as well as the reducing agent NADH, that are used in numerous other reactions. Its central importance to many biochemical pathways suggests that it was one of the earliest components of metabolism. Even though it is branded as a "cycle", it is not necessary for metabolites to follow only one specific route; at least three alternative segments of the citric acid cycle have been recognized.
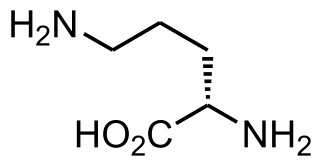
Proline (symbol Pro or P) is an organic acid classed as a proteinogenic amino acid (used in the biosynthesis of proteins), although it does not contain the amino group -NH
2 but is rather a secondary amine. The secondary amine nitrogen is in the protonated form (NH2+) under biological conditions, while the carboxyl group is in the deprotonated −COO− form. The "side chain" from the α carbon connects to the nitrogen forming a pyrrolidine loop, classifying it as a aliphatic amino acid. It is non-essential in humans, meaning the body can synthesize it from the non-essential amino acid L-glutamate. It is encoded by all the codons starting with CC (CCU, CCC, CCA, and CCG).
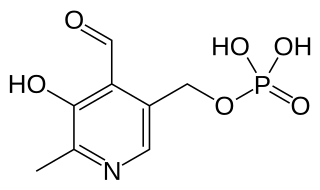
Pyridoxal phosphate (PLP, pyridoxal 5'-phosphate, P5P), the active form of vitamin B6, is a coenzyme in a variety of enzymatic reactions. The International Union of Biochemistry and Molecular Biology has catalogued more than 140 PLP-dependent activities, corresponding to ~4% of all classified activities. The versatility of PLP arises from its ability to covalently bind the substrate, and then to act as an electrophilic catalyst, thereby stabilizing different types of carbanionic reaction intermediates.
Biosynthesis, i.e., chemical synthesis occurring in biological contexts, is a term most often referring to multi-step, enzyme-catalyzed processes where chemical substances absorbed as nutrients serve as enzyme substrates, with conversion by the living organism either into simpler or more complex products. Examples of biosynthetic pathways include those for the production of amino acids, lipid membrane components, and nucleotides, but also for the production of all classes of biological macromolecules, and of acetyl-coenzyme A, adenosine triphosphate, nicotinamide adenine dinucleotide and other key intermediate and transactional molecules needed for metabolism. Thus, in biosynthesis, any of an array of compounds, from simple to complex, are converted into other compounds, and so it includes both the catabolism and anabolism of complex molecules. Biosynthetic processes are often represented via charts of metabolic pathways. A particular biosynthetic pathway may be located within a single cellular organelle, while others involve enzymes that are located across an array of cellular organelles and structures.
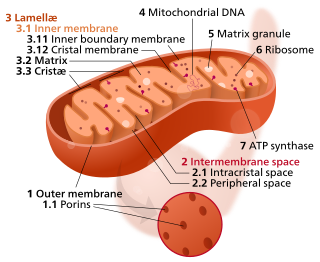
In the mitochondrion, the matrix is the space within the inner membrane. The word "matrix" stems from the fact that this space is viscous, compared to the relatively aqueous cytoplasm. The mitochondrial matrix contains the mitochondrial DNA, ribosomes, soluble enzymes, small organic molecules, nucleotide cofactors, and inorganic ions.[1] The enzymes in the matrix facilitate reactions responsible for the production of ATP, such as the citric acid cycle, oxidative phosphorylation, oxidation of pyruvate, and the beta oxidation of fatty acids.

Amino acid biosynthesis is the set of biochemical processes by which the amino acids are produced. The substrates for these processes are various compounds in the organism's diet or growth media. Not all organisms are able to synthesize all amino acids. For example, humans can synthesize 11 of the 20 standard amino acids. These 11 are called the non-essential amino acids.
Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.

Hyperprolinemia is a condition which occurs when the amino acid proline is not broken down properly by the enzymes proline oxidase or pyrroline-5-carboxylate dehydrogenase, causing a buildup of proline in the body.
In enzymology, an alanine racemase is an enzyme that catalyzes the chemical reaction
In enzymology, 4-aminobutyrate transaminase, also called GABA transaminase or 4-aminobutyrate aminotransferase, or GABA-T, is an enzyme that catalyzes the chemical reaction:
In enzymology, an acetylornithine transaminase (EC 2.6.1.11) is an enzyme that catalyzes the chemical reaction
In enzymology, a D-amino-acid transaminase is an enzyme that catalyzes the chemical reaction:
In enzymology, a succinylornithine transaminase (EC 2.6.1.81) is an enzyme that catalyzes the chemical reaction
Aspartate aminotransferase, mitochondrial is an enzyme that in humans is encoded by the GOT2 gene. Glutamic-oxaloacetic transaminase is a pyridoxal phosphate-dependent enzyme which exists in cytoplasmic and inner-membrane mitochondrial forms, GOT1 and GOT2, respectively. GOT plays a role in amino acid metabolism and the urea and Kreb's cycle. Also, GOT2 is a major participant in the malate-aspartate shuttle, which is a passage from the cytosol to the mitochondria. The two enzymes are homodimeric and show close homology. GOT2 has been seen to have a role in cell proliferation, especially in terms of tumor growth.

Aspartate aminotransferase, cytoplasmic is an enzyme that in humans is encoded by the GOT1 gene.

Delta-1-pyrroline-5-carboxylate synthetase (P5CS) is an enzyme that in humans is encoded by the ALDH18A1 gene. This gene is a member of the aldehyde dehydrogenase family and encodes a bifunctional ATP- and NADPH-dependent mitochondrial enzyme with both gamma-glutamyl kinase and gamma-glutamyl phosphate reductase activities. The encoded protein catalyzes the reduction of glutamate to delta1-pyrroline-5-carboxylate, a critical step in the de novo biosynthesis of proline, ornithine and arginine. Mutations in this gene lead to hyperammonemia, hypoornithinemia, hypocitrullinemia, hypoargininemia and hypoprolinemia and may be associated with neurodegeneration, cataracts and connective tissue diseases. Alternatively spliced transcript variants, encoding different isoforms, have been described for this gene. As reported by Bruno Reversade and colleagues, ALDH18A1 deficiency or dominant-negative mutations in P5CS in humans causes a progeroid disease known as De Barsy Syndrome.

Iminoglycinuria is an autosomal recessive disorder of renal tubular transport affecting reabsorption of the amino acid glycine, and the imino acids proline and hydroxyproline. This results in excess urinary excretion of all three acids.
Ornithine aminotransferase deficiency is an inborn error of ornithine metabolism, caused by decreased activity of the enzyme ornithine aminotransferase. Biochemically, it can be detected by elevated levels of ornithine in the blood. Clinically, it presents initially with poor night vision, which slowly progresses to total blindness. It is believed to be inherited in an autosomal recessive manner. Approximately 200 known cases have been reported in the literature. The incidence is highest in Finland, estimated at 1:50,000.
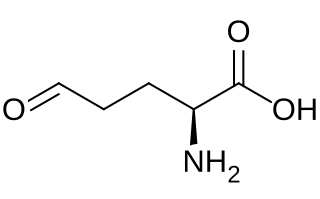
Glutamate-5-semialdehyde is a non-proteinogenic amino acid involved in both the biosynthesis and degradation of proline and arginine, as well as in the biosynthesis of antibiotics, such as carbapenems. It is synthesized by the reduction of glutamyl-5-phosphate by glutamate-5-semialdehyde dehydrogenase.
Arginine and proline metabolism is one of the central pathways for the biosynthesis of the amino acids arginine and proline from glutamate. The pathways linking arginine, glutamate, and proline are bidirectional. Thus, the net utilization or production of these amino acids is highly dependent on cell type and developmental stage. Altered proline metabolism has been linked to metastasis formation in breast cancer.
