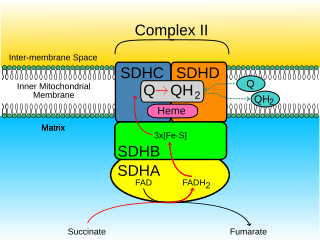
Succinate dehydrogenase [ubiquinone] cytochrome b small subunit, mitochondrial (CybS), also known as succinate dehydrogenase complex subunit D (SDHD), is a protein that in humans is encoded by the SDHD gene. Names previously used for SDHD were PGL and PGL1. Succinate dehydrogenase is an important enzyme in both the citric acid cycle and the electron transport chain. Hereditary PGL-PCC syndrome is caused by a parental imprint of the SDHD gene. Screening can begin by 6 years of age.
Succinate dehydrogenase complex subunit C, also known as succinate dehydrogenase cytochrome b560 subunit, mitochondrial, is a protein that in humans is encoded by the SDHC gene. This gene encodes one of four nuclear-encoded subunits that comprise succinate dehydrogenase, also known as mitochondrial complex II, a key enzyme complex of the tricarboxylic acid cycle and aerobic respiratory chains of mitochondria. The encoded protein is one of two integral membrane proteins that anchor other subunits of the complex, which form the catalytic core, to the inner mitochondrial membrane. There are several related pseudogenes for this gene on different chromosomes. Mutations in this gene have been associated with pheochromocytomas and paragangliomas. Alternatively spliced transcript variants have been described.
Coproporphyrinogen-III oxidase, mitochondrial is an enzyme that in humans is encoded by the CPOX gene. A genetic defect in the enzyme results in a reduced production of heme in animals. The medical condition associated with this enzyme defect is called hereditary coproporphyria.

Hyperprolinemia is a condition which occurs when the amino acid proline is not broken down properly by the enzymes proline oxidase or pyrroline-5-carboxylate dehydrogenase, causing a buildup of proline in the body.
In enzymology, proline dehydrogenase (PRODH) (EC 1.5.5.2, formerly EC 1.5.99.8) is an enzyme of the oxidoreductase family, active in the oxidation of L-proline to (S)-1-pyrroline-5-carboxylate during proline catabolism. The end product of this reaction is then further oxidized in a (S)-1-pyrroline-5-carboxylate dehydrogenase (P5CDH)-dependent reaction of the proline metabolism, or spent to produce ornithine, a crucial metabolite of ornithine and arginine metabolism. The systematic name of this enzyme class is L-proline:quinone oxidoreductase. Other names in common use include L-proline dehydrogenase, L-proline oxidase,and L-proline:(acceptor) oxidoreductase. It employs one cofactor, FAD, which requires riboflavin (vitamin B2).
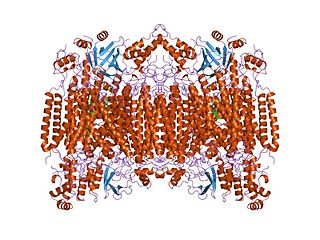
Cytochrome c oxidase I (COX1) also known as mitochondrially encoded cytochrome c oxidase I (MT-CO1) is a protein that is encoded by the MT-CO1 gene in eukaryotes. The gene is also called COX1, CO1, or COI. Cytochrome c oxidase I is the main subunit of the cytochrome c oxidase complex. In humans, mutations in MT-CO1 have been associated with Leber's hereditary optic neuropathy (LHON), acquired idiopathic sideroblastic anemia, Complex IV deficiency, colorectal cancer, sensorineural deafness, and recurrent myoglobinuria.

Cytochrome c oxidase II is a protein in eukaryotes that is encoded by the MT-CO2 gene. Cytochrome c oxidase subunit II, abbreviated COXII, COX2, COII, or MT-CO2, is the second subunit of cytochrome c oxidase. It is also one of the three mitochondrial DNA (mtDNA) encoded subunits of respiratory complex IV.
Cytochrome c oxidase subunit III (COX3) is an enzyme that in humans is encoded by the MT-CO3 gene. It is one of main transmembrane subunits of cytochrome c oxidase. It is also one of the three mitochondrial DNA (mtDNA) encoded subunits of respiratory complex IV. Variants of it have been associated with isolated myopathy, severe encephalomyopathy, Leber hereditary optic neuropathy, mitochondrial complex IV deficiency, and recurrent myoglobinuria.

A 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial is an enzyme that in humans is encoded by the BCKDHA gene.

Xaa-Pro dipeptidase, also known as prolidase, is an enzyme that in humans is encoded by the PEPD gene.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial (NDUFS4) also known as NADH-ubiquinone oxidoreductase 18 kDa subunit is an enzyme that in humans is encoded by the NDUFS4 gene. This gene encodes a nuclear-encoded accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase. Complex I removes electrons from NADH and passes them to the electron acceptor ubiquinone. Mutations in this gene can cause mitochondrial complex I deficiencies such as Leigh syndrome.

NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial (NDUFS1) is an enzyme that in humans is encoded by the NDUFS1 gene. The encoded protein, NDUFS1, is the largest subunit of complex I, located on the inner mitochondrial membrane, and is important for mitochondrial oxidative phosphorylation. Mutations in this gene are associated with complex I deficiency.

NADH dehydrogenase [ubiquinone] flavoprotein 2, mitochondrial (NDUFV2) is an enzyme that in humans is encoded by the NDUFV2 gene. The encoded protein, NDUFV2, is a subunit of complex I of the mitochondrial respiratory chain, which is located on the inner mitochondrial membrane and involved in oxidative phosphorylation. Mutations in this gene are implicated in Parkinson's disease, bipolar disorder, schizophrenia, and have been found in one case of early onset hypertrophic cardiomyopathy and encephalopathy.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 7, mitochondrial, also knowns as NADH-ubiquinone oxidoreductase 20 kDa subunit, Complex I-20kD (CI-20kD), or PSST subunit is an enzyme that in humans is encoded by the NDUFS7 gene. The NDUFS7 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain.

Pyrroline-5-carboxylate reductase 1, mitochondrial is an enzyme that in humans is encoded by the PYCR1 gene.

Delta-1-pyrroline-5-carboxylate dehydrogenase, mitochondrial is an enzyme that in humans is encoded by the ALDH4A1 gene.

Acyl-CoA dehydrogenase family member 9, mitochondrial is an enzyme that in humans is encoded by the ACAD9 gene. Mitochondrial Complex I Deficiency with varying clinical manifestations has been associated with mutations in ACAD9.

Cytochrome c oxidase assembly protein COX15 homolog (COX15), also known as heme A synthase, is a protein that in humans is encoded by the COX15 gene. This protein localizes to the inner mitochondrial membrane and involved in heme A biosynthesis. COX15 is also part of a three-component mono-oxygenase that catalyses the hydroxylation of the methyl group at position eight of the protoheme molecule. Mutations in this gene has been reported in patients with hypertrophic cardiomyopathy as well as Leigh syndrome, and characterized by delayed onset of symptoms, hypotonia, feeding difficulties, failure to thrive, motor regression, and brain stem signs.
22q11.2 duplication syndrome is a rare genetic disorder caused by a duplication of a segment at the end of chromosome 22.
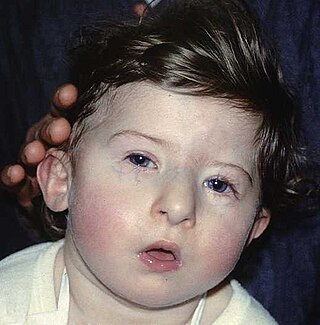
DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by a microdeletion on the long arm of chromosome 22. While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental delay, intellectual disability and cleft palate. Associated conditions include kidney problems, schizophrenia, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves' disease.




