
Biotin (or vitamin B7) is one of the B vitamins. It is involved in a wide range of metabolic processes, both in humans and in other organisms, primarily related to the utilization of fats, carbohydrates, and amino acids. The name biotin derives from the Greek word "bios" (to live) and the suffix "-in" (a suffix used in chemistry usually to indicate "forming").

Leucine (symbol Leu or L) is an essential amino acid that is used in the biosynthesis of proteins. Leucine is an α-amino acid, meaning it contains an α-amino group (which is in the protonated −NH3+ form under biological conditions), an α-carboxylic acid group (which is in the deprotonated −COO− form under biological conditions), and a side chain isobutyl group, making it a non-polar aliphatic amino acid. It is essential in humans, meaning the body cannot synthesize it: it must be obtained from the diet. Human dietary sources are foods that contain protein, such as meats, dairy products, soy products, and beans and other legumes. It is encoded by the codons UUA, UUG, CUU, CUC, CUA, and CUG.

β-Hydroxy β-methylbutyric acid (HMB), otherwise known as its conjugate base, β-hydroxyβ-methylbutyrate, is a naturally produced substance in humans that is used as a dietary supplement and as an ingredient in certain medical foods that are intended to promote wound healing and provide nutritional support for people with muscle wasting due to cancer or HIV/AIDS. In healthy adults, supplementation with HMB has been shown to increase exercise-induced gains in muscle size, muscle strength, and lean body mass, reduce skeletal muscle damage from exercise, improve aerobic exercise performance, and expedite recovery from exercise. Medical reviews and meta-analyses indicate that HMB supplementation also helps to preserve or increase lean body mass and muscle strength in individuals experiencing age-related muscle loss. HMB produces these effects in part by stimulating the production of proteins and inhibiting the breakdown of proteins in muscle tissue. No adverse effects from long-term use as a dietary supplement in adults have been found.
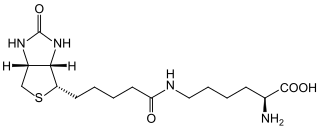
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.
3-Methylcrotonyl-CoA carboxylase deficiency also known as 3-Methylcrotonylglycinuria or BMCC deficiency is an inherited disorder in which the body is unable to process certain proteins properly. People with this disorder have inadequate levels of an enzyme that helps break down proteins containing the amino acid leucine. This condition affects an estimated 1 in 50,000 individuals worldwide.

β-Hydroxybutyric acid, also known as 3-hydroxybutyric acid or BHB, is an organic compound and a beta hydroxy acid with the chemical formula CH3CH(OH)CH2CO2H; its conjugate base is β-hydroxybutyrate, also known as 3-hydroxybutyrate. β-Hydroxybutyric acid is a chiral compound with two enantiomers: D-β-hydroxybutyric acid and L-β-hydroxybutyric acid. Its oxidized and polymeric derivatives occur widely in nature. In humans, D-β-hydroxybutyric acid is one of two primary endogenous agonists of hydroxycarboxylic acid receptor 2 (HCA2), a Gi/o-coupled G protein-coupled receptor (GPCR).
β-Hydroxy β-methylglutaryl-CoA (HMG-CoA), also known as 3-hydroxy-3-methylglutaryl coenzyme A, is an intermediate in the mevalonate and ketogenesis pathways. It is formed from acetyl CoA and acetoacetyl CoA by HMG-CoA synthase. The research of Minor J. Coon and Bimal Kumar Bachhawat in the 1950s at University of Illinois led to its discovery.
Propionyl-CoA is a coenzyme A derivative of propionic acid. It is composed of a 24 total carbon chain and its production and metabolic fate depend on which organism it is present in. Several different pathways can lead to its production, such as through the catabolism of specific amino acids or the oxidation of odd-chain fatty acids. It later can be broken down by propionyl-CoA carboxylase or through the methylcitrate cycle. In different organisms, however, propionyl-CoA can be sequestered into controlled regions, to alleviate its potential toxicity through accumulation. Genetic deficiencies regarding the production and breakdown of propionyl-CoA also have great clinical and human significance.

Enoyl-CoA hydratase (ECH) or crotonase is an enzyme EC 4.2.1.17 that hydrates the double bond between the second and third carbons on 2-trans/cis-enoyl-CoA:
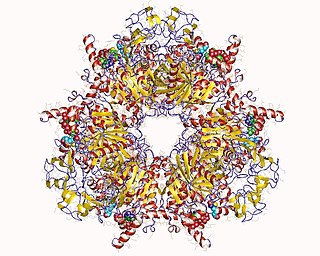
Propionyl-CoA carboxylase (EC 6.4.1.3, PCC) catalyses the carboxylation reaction of propionyl-CoA in the mitochondrial matrix. PCC has been classified both as a ligase and a lyase. The enzyme is biotin-dependent. The product of the reaction is (S)-methylmalonyl CoA.

3-Hydroxy-3-methylglutaryl-CoA lyase is an enzyme (EC 4.1.3.4 that in human is encoded by the HMGCL gene located on chromosome 1. It is a key enzyme in ketogenesis. It is a ketogenic enzyme in the liver that catalyzes the formation of acetoacetate from HMG-CoA within the mitochondria. It also plays a prominent role in the catabolism of the amino acid leucine.
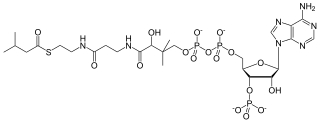
Isovaleryl-coenzyme A, also known as isovaleryl-CoA, is an intermediate in the metabolism of branched-chain amino acids.
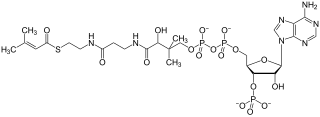
3-Methylcrotonyl-CoA or β-Methylcrotonyl-CoA is an intermediate in the metabolism of leucine.
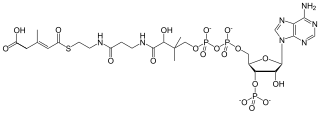
3-Methylglutaconyl-CoA (MG-CoA), also known as β-methylglutaconyl-CoA, is an intermediate in the metabolism of leucine. It is metabolized into HMG-CoA.

3-Methylglutaconyl-CoA hydratase, also known as MG-CoA hydratase and AUH, is an enzyme encoded by the AUH gene on chromosome 19. It is a member of the enoyl-CoA hydratase/isomerase superfamily, but it is the only member of that family that is able to bind to RNA. Not only does it bind to RNA, AUH has also been observed to be involved in the metabolic enzymatic activity, making it a dual-role protein. Mutations of this gene have been found to cause a disease called 3-Methylglutaconic Acuduria Type 1.
In enzymology, an isovaleryl-CoA dehydrogenase is an enzyme that catalyzes the chemical reaction
In enzymology, a biotin-[methylcrotonoyl-CoA-carboxylase] ligase is an enzyme that catalyzes the chemical reaction

In molecular biology, hydroxymethylglutaryl-CoA synthase or HMG-CoA synthase EC 2.3.3.10 is an enzyme which catalyzes the reaction in which acetyl-CoA condenses with acetoacetyl-CoA to form 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). This reaction comprises the second step in the mevalonate-dependent isoprenoid biosynthesis pathway. HMG-CoA is an intermediate in both cholesterol synthesis and ketogenesis. This reaction is overactivated in patients with diabetes mellitus type 1 if left untreated, due to prolonged insulin deficiency and the exhaustion of substrates for gluconeogenesis and the TCA cycle, notably oxaloacetate. This results in shunting of excess acetyl-CoA into the ketone synthesis pathway via HMG-CoA, leading to the development of diabetic ketoacidosis.

α-Ketoisocaproic acid (α-KIC) and its conjugate base, α-ketoisocaproate, are metabolic intermediates in the metabolic pathway for L-leucine. Leucine is an essential amino acid, and its degradation is critical for many biological duties. α-KIC is produced in one of the first steps of the pathway by branched-chain amino acid aminotransferase by transferring the amine on L-leucine onto alpha ketoglutarate, and replacing that amine with a ketone. The degradation of L-leucine in the muscle to this compound allows for the production of the amino acids alanine and glutamate as well. In the liver, α-KIC can be converted to a vast number of compounds depending on the enzymes and cofactors present, including cholesterol, acetyl-CoA, isovaleryl-CoA, and other biological molecules. Isovaleryl-CoA is the main compound synthesized from ɑ-KIC. α-KIC is a key metabolite present in the urine of people with Maple syrup urine disease, along with other branched-chain amino acids. Derivatives of α-KIC have been studied in humans for their ability to improve physical performance during anaerobic exercise as a supplemental bridge between short-term and long-term exercise supplements. These studies show that α-KIC does not achieve this goal without other ergogenicsupplements present as well. α-KIC has also been observed to reduce skeletal muscle damage after eccentrically biased resistance exercises in people who do not usually perform those exercises.
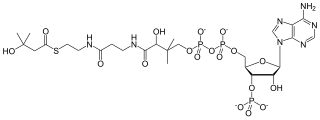
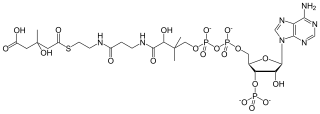
β-Hydroxy β-methylbutyryl-coenzyme A (HMB-CoA), also known as 3-hydroxyisovaleryl-CoA, is a metabolite of L-leucine that is produced in the human body. Its immediate precursors are β-hydroxy β-methylbutyric acid (HMB) and β-methylcrotonoyl-CoA (MC-CoA). It can be metabolized into HMB, MC-CoA, and HMG-CoA in humans.
