Achromatopsia, also known as rod monochromacy, is a medical syndrome that exhibits symptoms relating to five conditions, most notably monochromacy. Historically, the name referred to monochromacy in general, but now typically refers only to an autosomal recessive congenital color vision condition. The term is also used to describe cerebral achromatopsia, though monochromacy is usually the only common symptom. The conditions include: monochromatic color blindness, poor visual acuity, and day-blindness. The syndrome is also present in an incomplete form that exhibits milder symptoms, including residual color vision. Achromatopsia is estimated to affect 1 in 30,000 live births worldwide.

Retinitis pigmentosa (RP) is a member of a group of genetic disorders called inherited retinal dystrophy (IRD) that cause loss of vision. Symptoms include trouble seeing at night and decreasing peripheral vision. As peripheral vision worsens, people may experience "tunnel vision". Complete blindness is uncommon. Onset of symptoms is generally gradual and often begins in childhood.
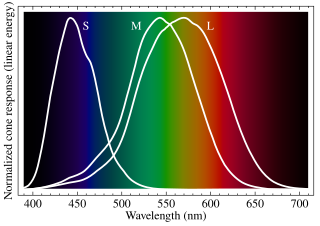
Cone cells or cones are photoreceptor cells in the retinas of vertebrates' eyes. They respond differently to light of different wavelengths, and the combination of their responses is responsible for color vision. Cones function best in relatively bright light, called the photopic region, as opposed to rod cells, which work better in dim light, or the scotopic region. Cone cells are densely packed in the fovea centralis, a 0.3 mm diameter rod-free area with very thin, densely packed cones which quickly reduce in number towards the periphery of the retina. Conversely, they are absent from the optic disc, contributing to the blind spot. There are about six to seven million cones in a human eye, with the highest concentration being towards the macula.
In visual physiology, adaptation is the ability of the retina of the eye to adjust to various levels of light. Natural night vision, or scotopic vision, is the ability to see under low-light conditions. In humans, rod cells are exclusively responsible for night vision as cone cells are only able to function at higher illumination levels. Night vision is of lower quality than day vision because it is limited in resolution and colors cannot be discerned; only shades of gray are seen. In order for humans to transition from day to night vision they must undergo a dark adaptation period of up to two hours in which each eye adjusts from a high to a low luminescence "setting", increasing sensitivity hugely, by many orders of magnitude. This adaptation period is different between rod and cone cells and results from the regeneration of photopigments to increase retinal sensitivity. Light adaptation, in contrast, works very quickly, within seconds.

Monochromacy is the ability of organisms to perceive only light intensity without respect to spectral composition. Organisms with monochromacy lack color vision and can only see in shades of grey ranging from black to white. Organisms with monochromacy are called monochromats. Many mammals, such as cetaceans, the owl monkey and the Australian sea lion are monochromats. In humans, monochromacy is one among several other symptoms of severe inherited or acquired diseases, including achromatopsia or blue cone monochromacy, together affecting about 1 in 30,000 people.

Nyctalopia, also called night-blindness, is a condition making it difficult or impossible to see in relatively low light. It is a symptom of several eye diseases. Night blindness may exist from birth, or be caused by injury or malnutrition. It can be described as insufficient adaptation to darkness.

Electroretinography measures the electrical responses of various cell types in the retina, including the photoreceptors, inner retinal cells, and the ganglion cells. Electrodes are placed on the surface of the cornea or on the skin beneath the eye to measure retinal responses. Retinal pigment epithelium (RPE) responses are measured with an EOG test with skin-contact electrodes placed near the canthi. During a recording, the patient's eyes are exposed to standardized stimuli and the resulting signal is displayed showing the time course of the signal's amplitude (voltage). Signals are very small, and typically are measured in microvolts or nanovolts. The ERG is composed of electrical potentials contributed by different cell types within the retina, and the stimulus conditions can elicit stronger response from certain components.

The Purkinje effect or Purkinje phenomenon is the tendency for the peak luminance sensitivity of the eye to shift toward the blue end of the color spectrum at low illumination levels as part of dark adaptation. In consequence, reds will appear darker relative to other colors as light levels decrease. The effect is named after the Czech anatomist Jan Evangelista Purkyně. While the effect is often described from the perspective of the human eye, it is well established in a number of animals under the same name to describe the general shifting of spectral sensitivity due to pooling of rod and cone output signals as a part of dark/light adaptation.
In the study of visual perception, scotopic vision is the vision of the eye under low-light conditions. The term comes from the Greek skotos, meaning 'darkness', and -opia, meaning 'a condition of sight'. In the human eye, cone cells are nonfunctional in low visible light. Scotopic vision is produced exclusively through rod cells, which are most sensitive to wavelengths of around 498 nm and are insensitive to wavelengths longer than about 640 nm. Under scotopic conditions, light incident on the retina is not encoded in terms of the spectral power distribution. Higher visual perception occurs under scotopic vision as it does under photopic vision.

A cone dystrophy is an inherited ocular disorder characterized by the loss of cone cells, the photoreceptors responsible for both central and color vision.
Progressive retinal atrophy (PRA) is a group of genetic diseases seen in certain breeds of dogs and, more rarely, cats. Similar to retinitis pigmentosa in humans, it is characterized by the bilateral degeneration of the retina, causing progressive vision loss culminating in blindness. The condition in nearly all breeds is inherited as an autosomal recessive trait, with the exception of the Siberian Husky (inherited as an X chromosome linked trait) and the Bullmastiff (inherited as an autosomal dominant trait). There is no treatment.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.
Stargardt disease is the most common inherited single-gene retinal disease. In terms of the first description of the disease, it follows an autosomal recessive inheritance pattern, which has been later linked to bi-allelic ABCA4 gene variants (STGD1). However, there are Stargardt-like diseases with mimicking phenotypes that are referred to as STGD3 and STGD4, and have a autosomal dominant inheritance due to defects with ELOVL4 or PROM1 genes, respectively. It is characterized by macular degeneration that begins in childhood, adolescence or adulthood, resulting in progressive loss of vision.

Congenital stationary night blindness (CSNB) is a rare non-progressive retinal disorder. People with CSNB often have difficulty adapting to low light situations due to impaired photoreceptor transmission. These patients may also have reduced visual acuity, myopia, nystagmus, fundus abnormalities, and strabismus. CSNB has two forms -- complete, also known as type-1 (CSNB1), and incomplete, also known as type-2 (CSNB2), which are distinguished by the involvement of different retinal pathways. In CSNB1, downstream neurons called bipolar cells are unable to detect neurotransmission from photoreceptor cells. CSNB1 can be caused by mutations in various genes involved in neurotransmitter detection, including NYX. In CSNB2, the photoreceptors themselves have impaired neurotransmission function; this is caused primarily by mutations in the gene CACNA1F, which encodes a voltage-gated calcium channel important for neurotransmitter release. CSNB has been identified in horses and dogs as the result of mutations in TRPM1, GRM6, and LRIT3 .
Rhodopsin kinase is a serine/threonine-specific protein kinase involved in phototransduction. This enzyme catalyses the following chemical reaction:

Glutamate receptor, metabotropic 6, also known as GRM6 or mGluR6, is a protein which in humans is encoded by the GRM6 gene.

S-arrestin is a protein that in humans is encoded by the SAG gene.

The mission of the Foundation Fighting Blindness is to fund research that will lead to the prevention, treatment and cures for the entire spectrum of retinal degenerative diseases, including retinitis pigmentosa, macular degeneration, Usher syndrome, Stargardt disease and related conditions. These diseases, which affect more than 10 million Americans and millions more throughout the world, often lead to severe vision loss or complete blindness.
The Mizuo–Nakamura Phenomenon is a phenomenon observed in Oguchi's disease. It was named after Gentaro Mizuo (1876–1913) and Bunpei Nakamura (1886–1969), Japanese ophthalmologists.
Occult macular dystrophy (OMD) is a rare inherited degradation of the retina, characterized by progressive loss of function in the most sensitive part of the central retina (macula), the location of the highest concentration of light-sensitive cells (photoreceptors) but presenting no visible abnormality. "Occult" refers to the degradation in the fundus being difficult to discern. The disorder is called "dystrophy" instead of "degradation" to distinguish its genetic origin from other causes, such as age. OMD was first reported by Y. Miyake et al. in 1989.
