
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosome abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Tay–Sachs disease is a genetic disorder that results in the destruction of nerve cells in the brain and spinal cord. The most common form is infantile Tay–Sachs disease, which becomes apparent around the age of three to six months of age, with the baby losing the ability to turn over, sit, or crawl. This is then followed by seizures, hearing loss, and inability to move, with death usually occurring by the age of three to five. Less commonly, the disease may occur later in childhood, adolescence, or adulthood. These forms tend to be less severe, but the juvenile form typically results in death by age 15.
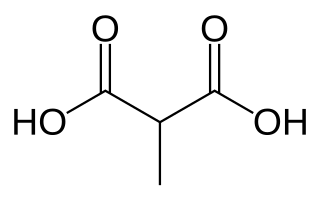
Methylmalonic acidemias, also called methylmalonic acidurias, are a group of inherited metabolic disorders, that prevent the body from properly breaking down proteins and fats. This leads to a buildup of a toxic level of methylmalonic acid in body liquids and tissues. Due to the disturbed branched-chain amino acids (BCAA) metabolism, they are among the classical organic acidemias.

Lesch–Nyhan syndrome (LNS) is a rare inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). This deficiency occurs due to mutations in the HPRT1 gene located on the X chromosome. LNS affects about 1 in 380,000 live births. The disorder was first recognized and clinically characterized by American medical student Michael Lesch and his mentor, pediatrician William Nyhan, at Johns Hopkins.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene–one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.
Pantothenate kinase-associated neurodegeneration (PKAN), formerly called Hallervorden–Spatz syndrome, is a genetic degenerative disease of the brain that can lead to parkinsonism, dystonia, dementia, and ultimately death. Neurodegeneration in PKAN is accompanied by an excess of iron that progressively builds up in the brain.
Pyruvate dehydrogenase deficiency is a rare neurodegenerative disorder associated with abnormal mitochondrial metabolism. PDCD is a genetic disease resulting from mutations in one of the components of the pyruvate dehydrogenase complex (PDC). The PDC is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria. The disorder shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.
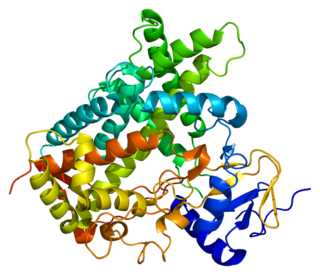
Cytochrome P450 2C19 is an enzyme protein. It is a member of the CYP2C subfamily of the cytochrome P450 mixed-function oxidase system. This subfamily includes enzymes that catalyze metabolism of xenobiotics, including some proton pump inhibitors and antiepileptic drugs. In humans, it is the CYP2C19 gene that encodes the CYP2C19 protein. CYP2C19 is a liver enzyme that acts on at least 10% of drugs in current clinical use, most notably the antiplatelet treatment clopidogrel (Plavix), drugs that treat pain associated with ulcers, such as omeprazole, antiseizure drugs such as mephenytoin, the antimalarial proguanil, and the anxiolytic diazepam.
In medical genetics, compound heterozygosity is the condition of having two or more heterogeneous recessive alleles at a particular locus that can cause genetic disease in a heterozygous state; that is, an organism is a compound heterozygote when it has two recessive alleles for the same gene, but with those two alleles being different from each other. Compound heterozygosity reflects the diversity of the mutation base for many autosomal recessive genetic disorders; mutations in most disease-causing genes have arisen many times. This means that many cases of disease arise in individuals who have two unrelated alleles, who technically are heterozygotes, but both the alleles are defective.

Methionine synthase reductase, also known as MSR, is an enzyme that in humans is encoded by the MTRR gene.

Neutral lipid storage disease is a congenital autosomal recessive disorder characterized by accumulation of triglycerides in the cytoplasm of leukocytes, muscle, liver, fibroblasts, and other tissues. It commonly occurs as one of two subtypes, cardiomyopathic neutral lipid storage disease (NLSD-M), or ichthyotic neutral lipid storage disease (NLSD-I) which is also known as Chanarin–Dorfman syndrome), which are characterized primarily by myopathy and ichthyosis, respectively. Normally, the ichthyosis that is present is typically non-bullous congenital ichthyosiform erythroderma which appears as white scaling.
Congenital generalized lipodystrophy is an extremely rare autosomal recessive condition, characterized by an extreme scarcity of fat in the subcutaneous tissues. It is a type of lipodystrophy disorder where the magnitude of fat loss determines the severity of metabolic complications. Only 250 cases of the condition have been reported, and it is estimated that it occurs in 1 in 10 million people worldwide.

Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding regions of genes in a genome. It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons—humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs. The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology.
DECIPHER is a web-based resource and database of genomic variation data from analysis of patient DNA. It documents submicroscopic chromosome abnormalities and pathogenic sequence variants, from over 25000 patients and maps them to the human genome using Ensembl or UCSC Genome Browser. In addition it catalogues the clinical characteristics from each patient and maintains a database of microdeletion/duplication syndromes, together with links to relevant scientific reports and support groups.
Non-allelic homologous recombination (NAHR) is a form of homologous recombination that occurs between two lengths of DNA that have high sequence similarity, but are not alleles.
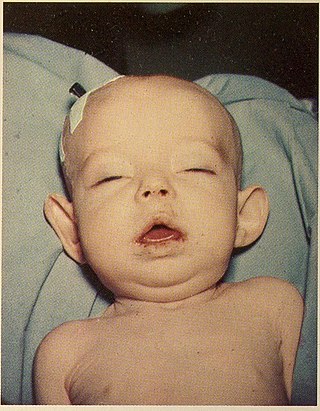
Donohue syndrome is an extremely rare and severe genetic disorder. Leprechaunism derives its name from the hallmark elvish features exhibited by the affected individuals. The disease is caused by a mutation in the INSR gene, which contains the genetic information for the formation of insulin receptors. As a result, affected individuals have either a decreased number of insulin receptors, or insulin receptor with greatly impaired functionality. The lack and impairment of insulin receptor functionality leads to an inability to regulate blood glucose levels through severe insulin resistance. This will ultimately lead to affected development of tissues and organs throughout the body. In addition to the physical abnormalities, leprechaunism is also characterized by endocrine system abnormalities that can lead to conditions such as hyperglycemia, hypoglycemia, hyperinsulemia, and the enlargement of certain sex organs such as the penis in males, and the clitoris in females.
Autism spectrum disorder (ASD) refers to a variety of conditions typically identified by challenges with social skills, communication, speech, and repetitive sensory-motor behaviors. The 11th International Classification of Diseases (ICD-11), released in January 2021, characterizes ASD by the associated deficits in the ability to initiate and sustain two-way social communication and restricted or repetitive behavior unusual for the individual's age or situation. Although linked with early childhood, the symptoms can appear later as well. Symptoms can be detected before the age of two and experienced practitioners can give a reliable diagnosis by that age. However, official diagnosis may not occur until much older, even well into adulthood. There is a large degree of variation in how much support a person with ASD needs in day-to-day life. This can be classified by a further diagnosis of ASD level 1, level 2, or level 3. Of these, ASD level 3 describes people requiring very substantial support and who experience more severe symptoms. ASD-related deficits in nonverbal and verbal social skills can result in impediments in personal, family, social, educational, and occupational situations. This disorder tends to have a strong correlation with genetics along with other factors. More research is identifying ways in which epigenetics is linked to autism. Epigenetics generally refers to the ways in which chromatin structure is altered to affect gene expression. Mechanisms such as cytosine regulation and post-translational modifications of histones. Of the 215 genes contributing, to some extent in ASD, 42 have been found to be involved in epigenetic modification of gene expression. Some examples of ASD signs are specific or repeated behaviors, enhanced sensitivity to materials, being upset by changes in routine, appearing to show reduced interest in others, avoiding eye contact and limitations in social situations, as well as verbal communication. When social interaction becomes more important, some whose condition might have been overlooked suffer social and other exclusion and are more likely to have coexisting mental and physical conditions. Long-term problems include difficulties in daily living such as managing schedules, hypersensitivities, initiating and sustaining relationships, and maintaining jobs.

A hereditary cancer syndrome is a genetic disorder in which inherited genetic mutations in one or more genes predispose the affected individuals to the development of cancer and may also cause early onset of these cancers. Hereditary cancer syndromes often show not only a high lifetime risk of developing cancer, but also the development of multiple independent primary tumors.
Elective genetic and genomic testing are DNA tests performed for an individual who does not have an indication for testing. An elective genetic test analyzes selected sites in the human genome while an elective genomic test analyzes the entire human genome. Some elective genetic and genomic tests require a physician to order the test to ensure that individuals understand the risks and benefits of testing as well as the results. Other DNA-based tests, such as a genealogical DNA test do not require a physician's order. Elective testing is generally not paid for by health insurance companies. With the advent of personalized medicine, also called precision medicine, an increasing number of individuals are undertaking elective genetic and genomic testing.

Salt and pepper developmental regression syndrome, also known as Amish infantile epileptic syndrome or GM3 deficiency syndrome, is a rare autosomal recessive progressive neurological disorder characterized by developmental delay, severe intellectual disability, seizures, and skin pigmentation irregularities. The clinical symptoms of this condition start manifesting soon after birth, during the newborn/neo-natal stage of life.



