Related Research Articles
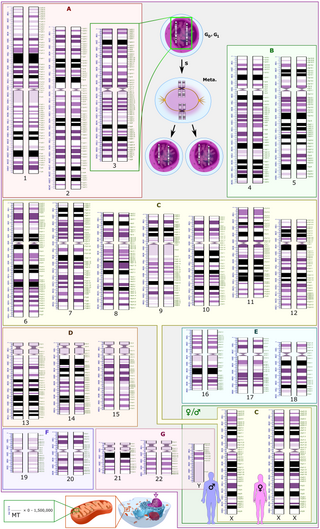
The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.
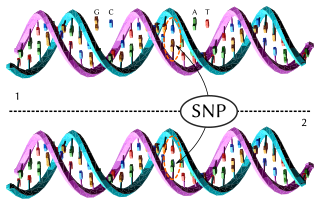
In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.
The International HapMap Project was an organization that aimed to develop a haplotype map (HapMap) of the human genome, to describe the common patterns of human genetic variation. HapMap is used to find genetic variants affecting health, disease and responses to drugs and environmental factors. The information produced by the project is made freely available for research.
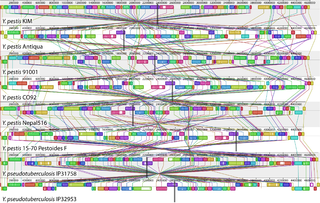
Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.

A DNA segment is identical by state (IBS) in two or more individuals if they have identical nucleotide sequences in this segment. An IBS segment is identical by descent (IBD) in two or more individuals if they have inherited it from a common ancestor without recombination, that is, the segment has the same ancestral origin in these individuals. DNA segments that are IBD are IBS per definition, but segments that are not IBD can still be IBS due to the same mutations in different individuals or recombinations that do not alter the segment.
In molecular biology, SNP array is a type of DNA microarray which is used to detect polymorphisms within a population. A single nucleotide polymorphism (SNP), a variation at a single site in DNA, is the most frequent type of variation in the genome. Around 335 million SNPs have been identified in the human genome, 15 million of which are present at frequencies of 1% or higher across different populations worldwide.
A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.
Human evolutionary genetics studies how one human genome differs from another human genome, the evolutionary past that gave rise to the human genome, and its current effects. Differences between genomes have anthropological, medical, historical and forensic implications and applications. Genetic data can provide important insights into human evolution.

In genomics, a genome-wide association study, is an observational study of a genome-wide set of genetic variants in different individuals to see if any variant is associated with a trait. GWA studies typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major human diseases, but can equally be applied to any other genetic variants and any other organisms.

The 1000 Genomes Project, launched in January 2008, was an international research effort to establish by far the most detailed catalogue of human genetic variation. Scientists planned to sequence the genomes of at least one thousand anonymous participants from a number of different ethnic groups within the following three years, using newly developed technologies which were faster and less expensive. In 2010, the project finished its pilot phase, which was described in detail in a publication in the journal Nature. In 2012, the sequencing of 1092 genomes was announced in a Nature publication. In 2015, two papers in Nature reported results and the completion of the project and opportunities for future research.
Expression quantitative trait loci (eQTLs) are genomic loci that explain variation in expression levels of mRNAs.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.

Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding regions of genes in a genome. It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons—humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs. The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology.
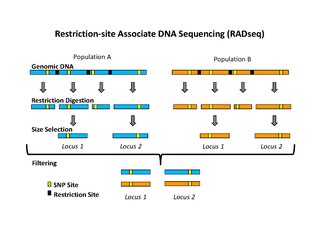
Restriction site associated DNA (RAD) markers are a type of genetic marker which are useful for association mapping, QTL-mapping, population genetics, ecological genetics and evolutionary genetics. The use of RAD markers for genetic mapping is often called RAD mapping. An important aspect of RAD markers and mapping is the process of isolating RAD tags, which are the DNA sequences that immediately flank each instance of a particular restriction site of a restriction enzyme throughout the genome. Once RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms mainly in form of single nucleotide polymorphisms (SNPs). Polymorphisms that are identified and genotyped by isolating and analyzing RAD tags are referred to as RAD markers. Although genotyping by sequencing presents an approach similar to the RAD-seq method, they differ in some substantial ways.
Imputation in genetics refers to the statistical inference of unobserved genotypes. It is achieved by using known haplotypes in a population, for instance from the HapMap or the 1000 Genomes Project in humans, thereby allowing to test for association between a trait of interest and experimentally untyped genetic variants, but whose genotypes have been statistically inferred ("imputed"). Genotype imputation is usually performed on SNPs, the most common kind of genetic variation.
Predictive genomics is at the intersection of multiple disciplines: predictive medicine, personal genomics and translational bioinformatics. Specifically, predictive genomics deals with the future phenotypic outcomes via prediction in areas such as complex multifactorial diseases in humans. To date, the success of predictive genomics has been dependent on the genetic framework underlying these applications, typically explored in genome-wide association (GWA) studies. The identification of associated single-nucleotide polymorphisms underpin GWA studies in complex diseases that have ranged from Type 2 Diabetes (T2D), Age-related macular degeneration (AMD) and Crohn's disease.
In the field of genetic sequencing, genotyping by sequencing, also called GBS, is a method to discover single nucleotide polymorphisms (SNP) in order to perform genotyping studies, such as genome-wide association studies (GWAS). GBS uses restriction enzymes to reduce genome complexity and genotype multiple DNA samples. After digestion, PCR is performed to increase fragments pool and then GBS libraries are sequenced using next generation sequencing technologies, usually resulting in about 100bp single-end reads. It is relatively inexpensive and has been used in plant breeding. Although GBS presents an approach similar to restriction-site-associated DNA sequencing (RAD-seq) method, they differ in some substantial ways.
Genome sequencing of endangered species is the application of Next Generation Sequencing (NGS) technologies in the field of conservation biology, with the aim of generating life history, demographic and phylogenetic data of relevance to the management of endangered wildlife.
References
- Population genomics: a bridge from evolutionary history to genetic medicine
- Black Iv, William C.; Baer, Charles F.; Antolin, Michael F.; Duteau, Nancy M. (2001). "POPULATION GENOMICS : Genome-Wide Sampling of Insect Populations". Annual Review of Entomology. 46: 441–469. doi:10.1146/annurev.ento.46.1.441. PMID 11112176.
- The Power and Promise of Population Genomics: From Genotyping to Genome Typing [ permanent dead link ]
- Schilling, M. P.; Wolf, P. G.; Duffy, A. M.; Rai, H. S.; Rowe, C. A.; Richardson, B. A.; Mock, K. E. (2014). "Genotyping-by-Sequencing for Populus Population Genomics: An Assessment of Genome Sampling Patterns and Filtering Approaches". PLOS ONE. 9 (4): 1–9. Bibcode:2014PLoSO...995292S. doi: 10.1371/journal.pone.0095292 . PMC 3991623 . PMID 24748384.