Turner syndrome (TS), commonly known as 45,X, or 45,XO,[note 1] is a chromosomal disorder in which cells have only one X chromosome or are partially missing an X chromosome (sex chromosomemonosomy) leading to the complete or partial deletion of the pseudoautosomal regions (PAR1, PAR2) in the affected X chromosome.[2][6][7] Most people have two sex chromosomes (XX or XY). The chromosomal abnormality is often present in just some cells, in which case it is known as Turner syndrome with mosaicism.[7][8] 45,X0 with mosaicism can occur in males or females,[9] but Turner syndrome without mosaicism only occurs in females.[2][6] Signs and symptoms vary among those affected but often include additional skin folds on the neck, arched palate, low-set ears, low hairline at the nape of the neck, short stature, and lymphedema of the hands and feet.[1] Typically, those affected do not develop menstrual periods or mammary glands without hormone treatment and are unable to reproduce without assistive reproductive technology. Small chin (micrognathia), loose folds of skin on the neck, slanted eyelids and prominent ears are found in Turner syndrome, though not all will show it. [1]Heart defects, Type II diabetes, and hypothyroidism occur in the disorder more frequently than average.[1] Most people with Turner syndrome have normal intelligence; however, many have problems with spatial visualization that may be needed in order to learn mathematics.[1] Ptosis (droopy eyelids) and conductive hearing loss also occur more often than average.[7]
Turner syndrome is caused by one X chromosome (45,X), a ring X chromosome, 45,X/46,XX mosaicism, or a small piece of the Y chromosome in what should be a X chromosome. They may have a total of 45 chromosomes or will not develop menstrual periods due to loss of ovarian function genes. Their karyotype often lacks Barr bodies due to lack of a second X or may have Xp deletions. it occurs during formation of the reproductive cells in a parent or in early cell division during development.[10][11] No environmental risks are known, and the mother's age does not play a role.[10][12] While most people have 46 chromosomes, people with Turner syndrome usually have 45 in some or all cells.[6] In cases of mosaicism, the symptoms are usually fewer, and possibly none occur at all.[13] Diagnosis is based on physical signs and genetic testing.[3]
No cure for Turner syndrome is known.[14] Treatment may help with symptoms.[14]Human growth hormone injections during childhood may increase adult height.[14]Estrogen replacement therapy can promote development of the breasts and hips.[14] Medical care is often required to manage other health problems with which Turner syndrome is associated.[14]
Turner syndrome occurs in between one in 2,000[4] and one in 5,000 females at birth.[5] All regions of the world and cultures are affected about equally.[10] Generally people with Turner syndrome have a shorter life expectancy, mostly due to heart problems and diabetes.[7] American endocrinologist Henry Turner first described the condition in 1938.[15] In 1964, it was determined to be due to a chromosomal abnormality.[15]
Presentation
Turner syndrome is associated with a number of physical features, including short stature, heart defects, webbed neck, micrognathia, amenorrhoea, and infertility.[16] The phenotype of Turner syndrome is affected by mosaicism, where cell lines with a single sex chromosome are combined with those with multiple. Individuals with mosaicism of 45,X0/46,XY may be phenotypically male, female, or ambiguous, while those with 45,X0/46,XX will be phenotypically female. Patients with 45,X0/46,XY do not receive the diagnosis of Turner syndrome if phenotypically male.[9] Around 40%–50% of cases of Turner syndrome are true "monosomy X" with a 45,X0 karyotype, while the remainder are mosaic for another cell line, most commonly 46,XX, or have other structural abnormalities of the X chromosome.[17] The classic features of Turner syndrome, while distinctive, may be rarer than previously thought; incidental diagnosis, such as in biobank samples or prenatal testing for older mothers, finds many girls and women with few traditional signs of Turner syndrome.[18][19]
Physiological
Stature
Height comparison for women with full and mosaic Turner's compared to trisomy X and the general population
Turner syndrome is associated with short stature. The mean adult height of women with Turner syndrome without growth hormone therapy is around 20cm (8in) shorter than the mean of women in the general population.[20]Mosaicism affects height in Turner syndrome; a large population sample drawn from the UK Biobank found women with 45,X0 karyotypes to have an average height of 145cm (4ft 9in), while those with 45,X0/46,XX karyotypes averaged 159cm (5ft 2+1⁄2in).[19][note 2] The strength of the association between Turner syndrome and short stature is such that idiopathic short stature alone is a major diagnostic indication.[17]
Growth delay in Turner syndrome does not begin at birth; most neonates with the condition have a birth weight in the lower end of the normal range. Height begins to lag in toddlerhood, with a delayed growth velocity becoming apparent as early as 18 months. Marked short stature becomes obvious in mid-childhood. In undiagnosed preadolescents and adolescents, growth delay may be mistaken for a side effect of delayed puberty and improperly treated.
Short stature in Turner syndrome and its counterpoint, tall stature in sex chromosome polysomy conditions such as Klinefelter syndrome, XYY syndrome, and trisomy X, is caused by the short-stature homeobox gene on the X and Y chromosomes. The absence of a copy of the SHOX gene in Turner's inhibits skeletal growth, resulting both in overall short stature and in a distinctive pattern of skeletal malformations including micrognathia (small chin), cubitus valgus (abnormal forearm angles), and shorter fingers.[23]
When Turner syndrome is diagnosed in early life, growth hormone therapy can decrease the degree of short stature.[17] Treatment with human growth hormone appears to increase expected adult height by approximately 7cm (3in) from an otherwise expected norm of 142cm (4ft 8in)–147cm (4ft 10in).[23][24][note 3] The effects of growth hormone therapy are at their strongest during the first year of treatment and taper off over time.[26] In some cases oxandrolone, a steroid with a relatively mild masculinizing effect, may be used alongside growth hormone. The addition of oxandrolone to a Turner syndrome treatment regimen adds around 2cm (1in) to the final height.[24][27] Oxandrolone is used particularly often in girls diagnosed later in their growth period, due to the reduced impact of growth hormone alone in this population. However, oxandrolone use runs the risk of delayed breast development, voice deepening, increased body hair, or clitoromegaly.[17]
Physical features
Girl with Turner syndrome
In addition to short stature, Turner syndrome is associated with a number of characteristic physical features. These include a short, webbed neck, low hairline, small chin and lower jaw, arched palate, and broad chest with widely-spaced nipples.[28]Lymphedema (swelling) of the hands and feet is common at birth and sometimes persistent throughout the lifespan.[29]
A number of the external manifestations of Turner syndrome are focused on the limbs, hands, and feet. Lymphedema at birth is one of the classic features of the syndrome; though it often resolves during toddlerhood, recurrence in later life is frequent, often without apparent cause. Cases where the retained X chromosome was inherited from the mother more often experience lymphedema than those where it was from the father. As a consequence of lymphedema's effects on nail anatomy, females with Turner syndrome frequently have small, hypoplastic, upturned nails. Their fingers are shorter and the hands are broad. Their feet are puffy, thicker, and swollen.[30] Shortened metacarpal bones, particularly the fourth metacarpal, are a frequent finding.[31] The body shape of individuals with Turner syndrome is frequently quite broad and stocky, as the growth deficiency is more pronounced in the length of bones than in their width. Scoliosis is common in Turner syndrome, and is seen in 40% of girls without growth hormone treatment.[32]
Facial features associated with Turner syndrome include broad, prominent ears, a low hairline at the nape of the neck, a webbed neck, a small chin with dental malocclusion, and downslanting palpebral fissures (the opening between the eyelids). These are thought to be related to lymphedema during the fetal period, specifically to the presence and resorption of excess fluids in the head and neck region.[17] Neck webbing is a particularly distinctive trait of Turner syndrome, leading to many neonatal diagnoses.[33] The underlying etiology of neck webbing is related to prenatal blood flow issues, and even in populations without Turner's has broad health consequences; the rate of congenital heart disease in webbed neck is 150-fold higher than in the general population, while the feature is also associated with reduced height and minor developmental impairments.[30] Some women with Turner syndrome have premature facial wrinkling.[17] Acne is less common in teenage girls and women with Turner syndrome, though the reasons why are unclear.[32]
A female infant with Turner syndrome. Notice the broad chest and small chin
Other physical features connected to the condition include long eyelashes, sometimes including an additional set of eyelashes, and unusual dermatoglyphics (fingerprints). Some women with Turner's report being unable to create fingerprint passwords due to hypoplastic dermatoglyphics.[17] Unusual dermatoglyphics are common to chromosome anomalies like Down Syndrome.[34] and in the case of Turner's may be a consequence of fetal lymphedema.[17]Keloid scars, or raised hypertrophic scars growing beyond the boundaries of the original wound, are potentially associated with Turner syndrome; however, the association is underresearched. Though traditional medical counselling on the topic urges conservatism about elective procedures such as ear piercing due to the risk of severe scarring, the actual consequences are unclear. Keloids in Turner syndrome are particularly frequent following surgical procedures to reduce neck webbing.[17][30] Turner syndrome has been associated with unusual patterns of hair growth, such as patches of short and long hair. Armpit and pubic hair is often sparse, while arm and leg hair is often thick. Though armpit hair is reduced in amount and thickness, the pattern in which it is implanted in the skin is as in men, rather than as in women.[32]
Cardiac
Bicuspid aortic valve
Approximately half of individuals with Turner syndrome have congenital heart defects. CHDs associated with Turner syndrome include bicuspid aortic valves (30%), coarctation of the aorta (15%), and abnormalities of the arteries in the head and neck.[17] A rare but potentially fatal complication of heart defects in Turner syndrome is aortic dissection, where the inner layer of the aorta tears open. Aortic dissection is six times as common in females with Turner syndrome as the general population and accounts for 8% of all deaths in the syndrome. The risk is substantially increased for individuals with bicuspid aortic valves, who make up 95% of patients with aortic dissection compared to 30% of all Turner's patients, and coarctation of the aorta, who make up 90% and 15% respectively.[35]
Coronary artery disease onsets earlier in life in women with Turner syndrome compared to controls, and mortality from cardiac events is increased. This is thought to be in part a function of the relationship between Turner syndrome and obesity; women with Turner syndrome have a higher percentage of body fat for their weight than control women, and their short stature makes weight control more difficult. Though coronary artery disease is frequently thought a disease of older adults, young women with Turner syndrome are more likely to develop the disease than their 46,XX peers. Treatment recommendations for women with Turner syndrome and coronary artery disease are as in the general population, but as Turner's increases the risk of type 2 diabetes, women with insulin resistance must weigh up the benefits of prophylactic or early statin treatment with the risk of Type II diabetes.[17]
Internal medicine
Turner syndrome is associated with a broad variety of health considerations, such as liver and kidney issues, obesity, diabetes, and hypertension.[17] Liver dysfunction is common in women with Turner syndrome, with 50–80% having elevated liver enzymes.[36]Non-alcoholic fatty liver disease is increased in prevalence in Turner syndrome, likely related in part to both conditions' associations with obesity. Hepatic vascular diseases are also seen in the syndrome as an aspect of Turner syndrome's broader vascular, aortic and cardiac impacts. Primary biliary cholangitis is more common in 45,X0 than 46,XX women. An unclear association exists between estrogen replacement therapy and liver dysfunction in Turner syndrome; some studies imply estrogen therapy worsens such conditions, while others imply improvement.[37]
Duplicated ureter
Kidney issues, such as horseshoe kidney, are sometimes observed in Turner syndrome.[36] Horseshoe kidney, where the kidneys are fused together in a U-shape, occurs in around 10% of Turner's cases compared to less than 0.5% of the general population. A missing kidney is observed in as many as 5% of individuals with Turner syndrome, compared to around 0.1% of the population. A duplicated ureter, where two ureters drain a single kidney, occurs in as much as 20–30% of the Turner syndrome population. Kidney malformations ( horseshoe kidney, etc.) in Turner syndrome may be more common in mosaicism than in the full 45,X karyotype.[38] Serious complications of the kidney anomalies associated with Turner syndrome are rare, although there is some risk of issues such as obstructive uropathy, where the flow of urine from the kidneys is blocked.[17]
Women with Turner syndrome are more likely than average to have high blood pressure; as many as 60% of women with the condition are hypertensive. Isolated diastolic hypertension often precedes systolic hypertension in the condition and may develop at a young age. Treatments for hypertension in Turner syndrome are as in the general population.[17]
Approximately 25–80% of women with Turner syndrome have some level of insulin resistance, and a minority develop type 2 diabetes. The risk of diabetes in Turner syndrome varies by karyotype and appears to be raised by specific deletions of the short arm of the X chromosome (Xp). One study found that while a relatively low 9% of women with Xq (long arm) deletions had type 2 diabetes, 18% of those with full 45,X0 karyotypes did, as well as 23% with Xp deletions. 43% of women with isochromosome Xq, who both lacked the short arm and had an additional copy of the long arm, developed type 2 diabetes.[36] Though part of the diabetes risk in Turner syndrome is a function of weight control, some is independent; age- and weight-matched women with non-Turner's ovarian failure have a lower diabetes risk than in Turner syndrome. Growth hormone treatment plays an unclear role in diabetes risk, as does estrogen supplementation.[17]
The association between Turner syndrome and other diseases, such as cancer, is unclear. Overall, women with Turner syndrome do not appear more likely to develop cancer than women with 46,XX karyotypes, but the specific pattern of what cancers are highest risk seems to differ. The risk of breast cancer appears lower in Turner's than in control women, perhaps due to decreased levels of estrogen. Neuroblastoma, a cancer of infancy and early childhood, has been reported in girls with Turner syndrome. Tumours of the nervous system, both the central nervous system and the peripheral nervous system, are overrepresented amongst cancers in Turner syndrome.[17] Furthermore, about 5.5% of Turner syndrome individuals have an extra, abnormal small supernumerary marker chromosome (sSMC) which consists of part of a Y chromosome. This partial Y chromosome-bearing sSMC may include the SRY gene located on the p arm of the Y chromosome at band 11.2 (notated as Yp11.2). This gene encodes the testis-determining factor protein (also known as sex-determining region Y protein). Turner syndrome individuals with this SRY gene-containing sSMC have a very real increased risk of developing gonadal tissue neoplasms such as gonadoblastomas and in situseminomas (also termed dysgerminomas to indicate that this tumor has the pathology of the testicular tumor, seminoma, but develops in ovaries[39]). In one study, 34 Turner syndrome girls without overt evidence of these tumors were found at preventative surgery to have a gonadoblastoma (7 cases), dysgerminoma (1 case), or non-specific in situ gonadal neoplasm (1 case).[40] Turner syndrome girls with this sSMC otherwise have typical features of the Turner syndrome except for a minority who also have hirsutism and/or clitoral enlargement.[41] Surgical removal of the gonads has been recommended to remove the threat of developing these sSMC-associated neoplasms.[41][42][43] Turner syndrome individuals with an sSMC that lacks the SRY gene are not at an increased risk of developing these cancers.[41][40]
Sensory
Hearing loss is common in Turner syndrome. Though at birth hearing is normal with good hearing abilities, chronic middle ear infections are frequent throughout childhood, which can cause permanent conductive hearing loss or deafness. Deaf individuals with Turner syndrome may use speech-reading, sign language, and cochlear implants. In adulthood, sensorineural hearing loss occurs more often than in 46,XX women and at younger ages; though differing thresholds of hearing loss make it difficult to compare between studies, younger adult women with Turner syndrome are routinely found to have disproportionate rates of hearing issues, with sometimes up to half of women in their 20s and 30s having difficulty hearing well.[44] This hearing loss is progressive; at the age of 40, women with Turner syndrome have equivalent hearing loss to 46,XX women aged 60, on average.[45] Cohort studies imply hearing loss may be more common in women who also have metabolic syndrome.[46] The high prevalence of sensorineural hearing loss in Turner syndrome appears to be related to SHOX deficiency.[23]
Ocular and visual disorders are also increased in prevalence in Turner syndrome. More than half of individuals with Turner syndrome have some form of eye disorder. This may be a consequence of shared genes on the X chromosome in both visual and ovarian development.[47] Nearly half of cases have hyperopia or myopia, usually mild. Strabismus, or misalignment of the eye, occurs in around one-fifth to one-third of girls with Turner syndrome. As with strabismus outside the Turner's context, it may be treated with glasses, patching, or surgical correction. Esotropia, where the eye turns inwards, is more common than exotropia, where it turns outwards.[47][48]Ptosis, or a drooping eyelid, is a common facial manifestation of Turner syndrome; it usually has no appreciable impact on vision, but severe cases may limit visual range and require surgical correction.[47] The rate of red-green colourblindness in Turner syndrome is 8%, the same as in XY males. This is due to red-green colourblindness being an X-linked recessive condition; in people with a single X chromosome, whether normal males or Turner females, only a single mutated X is necessary for symptoms. Red-green colourblindness may be underdiagnosed in the Turner context, as the rarity of the condition in females reduces the likelihood of screening, and practitioners may not connect that the karyotype of Turner syndrome increases the risk from the female baseline.[48]
Thyroid disease is common in Turner syndrome. Hypothyroidism is prevalent; 30%–50% of women with Turner syndrome have Hashimoto's disease, where the thyroid gland is slowly destroyed by an autoimmune reaction from the immune system. By age 50, half of women with Turner syndrome have subclinical or clinical hypothyroidism.[50]Hyperthyroidism and Graves' disease are also increased in prevalence, though more modestly. The Turner's presentation of hyperthyroidism is as in the general population, while the presentation of hypothyroidism is often atypical, with a mild early presentation yet a more severe progression.[51] Women with isochromosome Xq are more likely to develop autoimmune thyroid disease than women with other forms of Turner syndrome.[50]
The risk of irritable bowel syndrome is increased around fivefold in Turner syndrome, and that of ulcerative colitis around fourfold. Celiac disease is also increased in prevalence, with around 4–8% of Turner's patients having comorbid celiac disease compared to 0.5–1% of the general population. Diagnosis of such conditions is difficult due to their nonspecific early symptoms. In the Turner's context, diagnosis may in particular be missed due to growth delay; such conditions cause growth delay and failure to thrive when they onset in childhood, but as girls with Turner syndrome already have such delay, symptoms may be overlooked and ascribed to the original condition.[49]
Alopecia areata, or recurrent patchy hair loss, is three times as common in Turner syndrome as the general population. Alopecia in the Turner syndrome context is frequently treatment-resistant, also seen in other chromosome aneuploidies such as Down syndrome. Psoriasis is common in Turner syndrome, although the precise prevalence is unclear. Turner's psoriasis may be related to growth hormone treatment, as psoriasis as a side effect of such therapies has been reported in patients without the karyotype. Psoriasis may progress to psoriatic arthritis, and this progression may be more common in Turner syndrome. Vitiligo has been reported in conjunction with Turner syndrome, but the risk is unclear and may be a side effect of increased clinical attention to autoimmune disease in this population.[32]
Puberty
Histopathology of ovarian tissue in mosaic (A and B) and full (C) Turner syndrome
Puberty is delayed or absent in Turner syndrome. A 2019 literature review found that 13% of women with a 45,X0 karyotype could expect to experience spontaneous thelarche (breast development), while 9% would undergo spontaneous menarche (beginning of menstruation). These numbers were higher in women with mosaic Turner's; 63% with 45,X0/46,XX karyotypes experienced spontaneous thelarche and 39% spontaneous menarche, while 88% with 45,X0/47,XXX (the presence of a trisomy X cell line) experienced spontaneous thelarche and 66% spontaneous menarche. Unexpectedly, women with Y-chromosome cells also had increased rates of thelarche and menarche compared to the 45,X baseline, at 41% and 19%. However, few women with trisomy X or Y-chromosome cell lines were covered in the review, impeding extrapolation from these results.[52] 6% of women with Turner syndrome have regular menstrual cycles; the rest experience primary or secondary amenorrhea or other menstrual dysfunction.[53]:84
In girls with Turner syndrome who do not experience spontaneous puberty, exogenous estrogen is used to induce and maintain feminization. Estrogen replacement is recommended to begin at around age 11–12, although some parents prefer to delay the induction of puberty in girls with lower social and emotional preparedness. The dose of estrogen in induced puberty begins at 10% of adult estrogen levels and is steadily increased at six-month intervals, with a full adult dose attained two to three years after the beginning of treatment. Estrogen replacement may interfere with growth hormone therapy, due to the closing effects of estrogen on growth plates; individuals must weigh up their preferences for taller height versus greater feminization.[53]:97–103
Fertility
Women with Turner syndrome are at extremely high risk for primary ovarian insufficiency (POI) and infertility. Although about 70–80% have no spontaneous pubertal development and 90% experience primary amenorrhea, the remainder may possess a small residual of ovarian follicles at birth or early childhood.[54]
Early in gestation, fetuses with Turner syndrome have a normal number of gametes in their developing ovaries, but this starts decreasing rapidly as early as 18 weeks of pregnancy; by birth, girls with the condition have markedly reduced follicular counts.[55] Women with Turner syndrome who wish to raise families but are incapable of conception with their own oocytes have the options of adoption or of pregnancy with donor eggs; the latter has a comparable success rate to donor pregnancy in women with 46,XX karyotypes.[17]
Usually, estrogen replacement therapy is used to spur the growth of secondary sexual characteristics at the time when puberty should onset. While very few women with Turner syndrome menstruate spontaneously, estrogen therapy requires a regular shedding of the uterine lining ("withdrawal bleeding") to prevent its overgrowth. Withdrawal bleeding can be induced monthly, like menstruation, or less often, usually every three months, if the patient desires. Estrogen therapy does not make a woman with nonfunctional ovaries fertile, but it plays an important role in assisted reproduction; the health of the uterus must be maintained with estrogen if an eligible woman with Turner Syndrome wishes to use IVF (using donated oocytes).[citation needed]
Especially in mosaic cases of Turner syndrome that contains Y-chromosome (e.g., 45,X/46,XY) due to the risk of development of ovarian malignancy (most common is gonadoblastoma) gonadectomy is recommended.[57][58] Turner syndrome is characterized by primary amenorrhoea, premature ovarian failure (hypergonadotropic hypogonadism), streak gonads and infertility (however, technology (especially oocyte donation) provides the opportunity of pregnancy in these patients). Failure to develop secondary sex characteristics (sexual infantilism) is typical.[citation needed]
Cognition
Neurodevelopmental
Individuals with Turner syndrome have normal intelligence. Verbal IQ is usually higher than performance IQ; one review of thirteen studies found an average verbal IQ of 101 compared to an average performance IQ of 89.[59]
People with Turner syndrome demonstrate relative strengths in verbal skills, but may exhibit weaker nonverbal skills – particularly in arithmetic, select visuospatial skills, and processing speed. They have difficulties with directional sense, visualization of three-dimensional shapes, properties of shapes, and symmetry and may have dyscalculia. [60] Turner syndrome does not typically cause intellectual disability or impair cognition. However, learning difficulties are common among women with Turner syndrome, particularly a specific difficulty in perceiving spatial relationships, such as nonverbal learning disorder. This may also manifest itself as a difficulty with motor control or with mathematics.[61] While it is not correctable, in most cases it does not cause difficulty in daily living. Most Turner syndrome patients are employed as adults and lead productive lives.
Also, a rare variety of Turner syndrome, known as "Ring-X Turner syndrome", has about a 60% association with intellectual disability[clarification needed]. This variety accounts for around 2–4% of all Turner syndrome cases.[62]
Psychological
Social difficulties appear to be an area of vulnerability for TS girls.[63] Counseling affected individuals and their families about the need to carefully develop social skills and relationships may prove useful in advancing social adaptation. Women with Turner syndrome may experience adverse psychosocial outcomes which can be improved through early intervention and the provision of appropriate psychological and psychiatric care. Genetic, hormonal, and medical problems associated with Turner syndrome are likely to affect psychosexual development of female adolescent patients, and thus influence their psychological functioning, behavior patterns, social interactions, and learning ability. Although Turner syndrome constitutes a chronic medical condition, with possible physical, social, and psychological complications in a woman's life, hormonal and estrogen replacement therapy, and assisted reproduction, are treatments that can be helpful for Turner syndrome patients and improve their quality of life.[64] Research shows a possible association between age at diagnosis and increased substance use and depressive symptoms.[65]
Prenatal
Despite the excellent postnatal prognosis, 99% of Turner syndrome conceptions are thought to end in miscarriage or stillbirth,[66] and as many as 15% of all spontaneous abortions have the 45,X karyotype.[67][68] Among cases that are detected by routine amniocentesis or chorionic villus sampling, one study found that the prevalence of Turner syndrome among tested pregnancies was 5.58 and 13.3 times higher, respectively, than among live neonates in a similar population.[69]
Cause
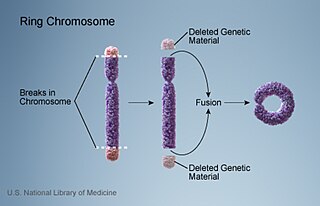
Turner syndrome is caused by the complete or partial absence of one copy of the X chromosome in some or all the cells. The abnormal cells may have only one X (monosomy) (45,X) or they may be affected by one of several types of partial monosomy like a deletion of the short p arm of one X chromosome (46,X,del(Xp)) or the presence of an isochromosome with two q arms (46,X,i(Xq))[70] Turner syndrome has distinct features due to the lack of pseudoautosomal regions, which are typically spared from X-inactivation.[7] In mosaic individuals, cells with X monosomy (45,X) may occur along with cells that are normal (46,XX), cells that have partial monosomies, or cells that have a Y chromosome (46,XY).[70] The presence of mosaicism is estimated to be relatively common in affected individuals (67–90%).[70]
The (46,X,i(Xq) isochromosome in the Turner syndrome is classified as a small supernumerary marker chromosome (sSMC). Two of the types of sSMCs in this syndrome contain parts of the genetic material from either an X or, much less frequently, Y chromosome and may or may not contain an XIST gene.[71] Turner syndrome females with (46,X,i(Xq) sSMC consisting of a partial X chromosome that does not contain the XIST gene express at least some of this sSMC's genetic material and therefore contain excesses of this material. In consequence, they have a more serious form of the Turner syndrome that ranges form moderately severe to extremely severe. The extremely severe cases have anencephaly (absence of a major portion of the brain, skull, and scalp), agenesis of the corpus callosum (lack of the thick tract of nerve fibers that connect the left and right cerebral hemispheres), and complex heart deformities. Individuals with Turner syndrome that have partial X chromosome containing(46,X,i(Xq) sSMCs that have the XIST gene do not express this sSMC's genetic material and do not have the more severe manifestations of the syndrome.[72]
Inheritance
In the majority of cases where monosomy occurs, the X chromosome comes from the mother.[73] This may be due to a nondisjunction in the father. Meiotic errors that lead to the production of X with p arm deletions or abnormal Y chromosomes are also mostly found in the father.[74]Isochromosome X or ring chromosome X on the other hand are formed equally often by both parents.[74] Overall, the functional X chromosome usually comes from the mother.
In most cases, Turner syndrome is a sporadic event, and for the parents of an individual with Turner syndrome the risk of recurrence is not increased for subsequent pregnancies. Rare exceptions may include the presence of a balanced translocation of the X chromosome in a parent, or where the mother has 45,X mosaicism restricted to her germ cells.[75]
Diagnosis
Prenatal
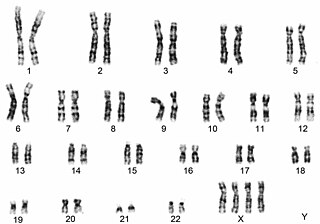
45,X karyotype, showing an unpaired X at the lower right
Usually, fetuses with Turner syndrome can be identified by abnormal ultrasound findings (i.e., heart defect, kidney abnormality, cystic hygroma, ascites). In a study of 19 European registries, 67.2% of prenatally diagnosed cases of Turner syndrome were detected by abnormalities on ultrasound. 69.1% of cases had one anomaly present, and 30.9% had two or more anomalies.[76]
An increased risk of Turner syndrome may also be indicated by abnormal triple or quadruple maternal serum screen. The fetuses diagnosed through positive maternal serum screening are more often found to have a mosaic karyotype than those diagnosed based on ultrasonographic abnormalities, and conversely, those with mosaic karyotypes are less likely to have associated ultrasound abnormalities.[76]
Postnatal
Turner syndrome can be diagnosed postnatally at any age. Often, it is diagnosed at birth due to heart problems, an unusually wide neck or swelling of the hands and feet. However, it is also common for it to go undiagnosed for several years, often until the girl reaches the age of puberty and fails to develop typically (the changes associated with puberty do not occur). In childhood, a short stature can be indicative of Turner syndrome.[77]
A test called a karyotype, also known as a chromosome analysis, analyzes the chromosomal composition of the individual. This is the test of choice to diagnose Turner syndrome.[78][79]
Treatment
As a chromosomal condition, there is no cure for Turner syndrome. However, much can be done to minimize the symptoms.[80] While most of the physical findings are harmless, significant medical problems can be associated with the syndrome. Most of these significant conditions are treatable with surgery and other therapies including hormonal therapy.[81]
Synthetic Growth hormone, either alone or with a low dose of androgen, will increase height by several inches and probably final adult height. Growth hormone is approved by the U.S. Food and Drug Administration for treatment of Turner syndrome and is covered by many insurance plans. It can make Turner females achieve a more normal height[80][82] There is evidence that this is effective, even in toddlers.[83] A 2019 systematic review comparing effects of adding oxandrolone to growth hormone treatment to growth hormone alone has found moderate-quality evidence that the addition of oxandrolone leads to an increase in final adult height of girls with Turner syndrome.[84] When the same review assessed the effects of adding Oxandrolone to growth hormone treatment on speech, cognition and psychological status, the results were inconclusive due to very-low quality evidence.[84]
Estrogen replacement therapy such as the birth control pill, has been used since the condition was described in 1938 to promote development of secondary sexual characteristics. Estrogens are crucial for maintaining good bone integrity, cardiovascular health and tissue health.[80] Women with Turner syndrome who do not have spontaneous puberty and who are not treated with estrogen are at high risk for osteoporosis and heart conditions.
Modern reproductive technologies have also been used to help women with Turner syndrome become pregnant if they desire. For example, a donor egg can be used to create an embryo, which is carried by the Turner syndrome woman.[80]
Uterine maturity is positively associated with years of estrogen use, history of spontaneous menarche, and negatively associated with the lack of current hormone replacement therapy.[85][86]
Epidemiology
Turner syndrome occurs in between one in 2,000[4] and one in 5,000 females at birth.[5]
Approximately 99 percent of fetuses with Turner syndrome spontaneously terminate during the first trimester.[87] Turner syndrome accounts for about 10 percent of the total number of spontaneous abortions in the United States.[57]
History
The syndrome is named after Henry Turner, an American endocrinologist, who described it in 1938.[88][89] n Europe, it is often called Ullrich–Turner syndrome and was sometimes called Bonnevie–Ullrich syndrome although the latter term is rarely used today.[90] Both syndrome names acknowledge(d) that earlier cases had also been described 1930 by European doctors Kristine Bonnevie and Otto Ullrich. In Russian and Soviet literature, it is called Shereshevsky–Turner syndrome to acknowledge that the condition was first described as hereditary in 1925 by the Soviet endocrinologist Nikolai Shereshevsky[ru], who believed that it was due to the underdevelopment of the gonads and the anterior pituitary gland and was combined with congenital malformations of internal development.[91]
The first published report of a female with a 45,X karyotype was in 1959 by Charles Ford and colleagues in Harwell near Oxford, and Guy's Hospital in London.[92] It was found in a 14-year-old girl with signs of Turner syndrome.[92]
Cultural & social impacts
In many cultures, the symptoms of Turner Syndrome—particularly short stature and infertility—carried social stigmas. Historically, women who were unable to conceive were often marginalized or considered undesirable as wives. While the understanding of TS has improved, the psychological impact of infertility remains a significant concern for many women with the condition.[93] In modern times, advocacy groups have played a vital role in raising awareness and offering support to those with TS, helping to reduce stigma and provide better access to medical care.[93]
↑ As comparison, the average adult height for women in the Anglosphere is around 162cm (5ft 4in).[21][22]
↑ Expected adult height for untreated women with Turner syndrome ranges from 143cm (4ft 8+1⁄2in) in the United States and most of Western Europe, to 140cm (4ft 7in) in Argentina, to 147cm (4ft 10in) in Scandinavia.[25]
Related Research Articles
Amenorrhea or amenorrhoea is the absence of a menstrual period in a female who has reached reproductive age. Physiological states of amenorrhoea are seen, most commonly, during pregnancy and lactation (breastfeeding). Outside the reproductive years, there is absence of menses during childhood and after menopause.
Nondisjunction is the failure of homologous chromosomes or sister chromatids to separate properly during cell division (mitosis/meiosis). There are three forms of nondisjunction: failure of a pair of homologous chromosomes to separate in meiosis I, failure of sister chromatids to separate during meiosis II, and failure of sister chromatids to separate during mitosis. Nondisjunction results in daughter cells with abnormal chromosome numbers (aneuploidy).
XY complete gonadal dysgenesis, also known as Swyer syndrome, is a type of defect hypogonadism in a person whose karyotype is 46,XY. Though they typically have normal vulvas, the person has underdeveloped gonads, fibrous tissue termed "streak gonads", and if left untreated, will not experience puberty. The cause is a lack or inactivation of an SRY gene which is responsible for sexual differentiation. Pregnancy is sometimes possible in Swyer syndrome with assisted reproductive technology. The phenotype is usually similar to Turner syndrome (45,X0) due to a lack of X inactivation. The typical medical treatment is hormone replacement therapy. The syndrome was named after Gerald Swyer, an endocrinologist based in London.
Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 15. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.
Oxandrolone is an androgen and synthetic anabolic steroid (AAS) medication to help promote weight gain in various situations, to help offset protein catabolism caused by long-term corticosteroid therapy, to support recovery from severe burns, to treat bone pain associated with osteoporosis, to aid in the development of girls with Turner syndrome, and for other indications. It is taken by mouth. It was sold under the brand names Oxandrin and Anavar, among others.
McCune–Albright syndrome is a complex genetic disorder affecting the bone, skin and endocrine systems. It is a mosaic disease arising from somatic activating mutations in GNAS, which encodes the alpha-subunit of the Gs heterotrimeric G protein.
Primary ovarian insufficiency (POI), also called premature ovarian insufficiency and premature ovarian failure, is the partial or total loss of reproductive and hormonal function of the ovaries before age 40 because of follicular dysfunction or early loss of eggs. POI can be seen as part of a continuum of changes leading to menopause that differ from age-appropriate menopause in the age of onset, degree of symptoms, and sporadic return to normal ovarian function. POI affects approximately 1 in 10,000 women under age 20, 1 in 1,000 women under age 30, and 1 in 100 of those under age 40. A medical triad for the diagnosis is amenorrhea, hypergonadotropism, and hypoestrogenism.
XX gonadal dysgenesis is a type of female hypogonadism in which the ovaries do not function to induce puberty in a person assigned female at birth, whose karyotype is 46,XX. Individuals with XX gonadal dysgenesis have normal-appearing external genitalia as well as Müllerian structures. Due to the nearly absent or nonfunctional streak ovaries, the individual is low in estrogen levels (hypoestrogenic) and has high levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH), hormones that cycle in the reproductive system. As a result, the diagnosis often occurs after a concern for delayed puberty or amenorrhea. Treatment generally involves hormone replacement therapy with estrogen and progesterone.
Gonadal dysgenesis is classified as any congenital developmental disorder of the reproductive system characterized by a progressive loss of primordial germ cells on the developing gonads of an embryo. One type of gonadal dysgenesis is the development of functionless, fibrous tissue, termed streak gonads, instead of reproductive tissue. Streak gonads are a form of aplasia, resulting in hormonal failure that manifests as sexual infantism and infertility, with no initiation of puberty and secondary sex characteristics.
Disorders of sex development (DSDs), also known as differences in sex development or variations in sex characteristics (VSC), are congenital conditions affecting the reproductive system, in which development of chromosomal, gonadal, or anatomical sex is atypical. DSDs is a clinical term used in some medical settings for what are otherwise referred to as intersex traits. The term was first introduced in 2006 and has not been without controversy.
A gonadoblastoma is a complex neoplasm composed of a mixture of gonadal elements, such as large primordial germ cells, immature Sertoli cells or granulosa cells of the sex cord, and gonadal stromal cells. Gonadoblastomas are by definition benign, but more than 50% have a co-existing dysgerminoma which is malignant, and an additional 10% have other more aggressive malignancies, and as such are often treated as malignant.
Klinefelter syndrome (KS), also known as 47,XXY, is a chromosome anomaly where a male has an extra X chromosome. These complications commonly include infertility and small, poorly functioning testicles. These symptoms are often noticed only at puberty, although this is one of the most common chromosomal disorders, occurring in one to two per 1,000 live births. It is named after American endocrinologist Harry Klinefelter, who identified the condition in the 1940s, along with his colleagues at Massachusetts General Hospital.
Wilson-Turner syndrome (WTS), also known as mental retardation X linked syndromic 6 (MRXS6), and mental retardation X linked with gynecomastia and obesity is a congenital condition characterized by intellectual disability and associated with childhood-onset obesity. It is found to be linked to the X chromosome and caused by a mutation in the HDAC8 gene, which is located on the q arm at locus 13.1. Individuals with Wilson–Turner syndrome have a spectrum of physical characteristics including dysmorphic facial features, hypogonadism, and short stature. Females generally have milder phenotypes than males. This disorder affects all demographics equally and is seen in less than one in one million people.
Hypergonadotropic hypogonadism (HH), also known as primary or peripheral/gonadal hypogonadism or primary gonadal failure, is a condition which is characterized by hypogonadism which is due to an impaired response of the gonads to the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and in turn a lack of sex steroid production. As compensation and the lack of negative feedback, gonadotropin levels are elevated. Individuals with HH have an intact and functioning hypothalamus and pituitary glands so they are still able to produce FSH and LH. HH may present as either congenital or acquired, but the majority of cases are of the former nature. HH can be treated with hormone replacement therapy.
45,X/46,XY mosaicism, also known as X0/XY mosaicism and mixed gonadal dysgenesis, is a mutation of sex development in humans associated with sex chromosome aneuploidy and mosaicism of the Y chromosome. It is a fairly rare chromosomal disorder at birth, with an estimated incidence rate of about 1 in 15,000 live births. Mosaic loss of the Y chromosome in previously non-mosaic men grows increasingly common with age.
Ring chromosome 15 is a condition that arises when chromosome 15 fuses to form a ring chromosome. Usually, ring chromosome 15 forms due to the modification or deletion of genetic information on chromosome 15 in the preliminary stages of embryonic development, but it can rarely also be inherited.
Tetrasomy X, also known as 48,XXXX, is a chromosomal disorder in which a female has four, rather than two, copies of the X chromosome. It is associated with intellectual disability of varying severity, characteristic "coarse" facial features, heart defects, and skeletal anomalies such as increased height, clinodactyly, and radioulnar synostosis. Tetrasomy X is a rare condition, with few medically recognized cases; it is estimated to occur in approximately 1 in 50,000 females.
Sexual anomalies, also known as sexual abnormalities, are a set of clinical conditions due to chromosomal, gonadal and/or genitalia variation. Individuals with congenital (inborn) discrepancy between sex chromosome, gonadal, and their internal and external genitalia are categorised as individuals with a disorder of sex development (DSD). Afterwards, if the family or individual wishes, they can partake in different management and treatment options for their conditions.
Pentasomy X, also known as 49,XXXXX, is a chromosomal disorder in which a female has five, rather than two, copies of the X chromosome. Pentasomy X is associated with short stature, intellectual disability, characteristic facial features, heart defects, skeletal anomalies, and pubertal and reproductive abnormalities. The condition is exceptionally rare, with an estimated prevalence between 1 in 85,000 and 1 in 250,000.
Trisomy X, also known as triple X syndrome and characterized by the karyotype 47,XXX, is a chromosome disorder in which a female has an extra copy of the X chromosome. It is relatively common and occurs in 1 in 1,000 females, but is rarely diagnosed; fewer than 10% of those with the condition know they have it.
1 2 3 "Turner Syndrome: Condition Information". Eunice Kennedy Shriver National Institute of Child Health and Human Development. 30 November 2012. Archived from the original on 29 March 2015. Retrieved 15 March 2015.
↑ "What causes Turner syndrome?". Eunice Kennedy Shriver National Institute of Child Health and Human Development. 30 November 2012. Archived from the original on 2 April 2015. Retrieved 15 March 2015.
↑ Gunther DF, Eugster E, Zagar AJ, Bryant CG, Davenport ML, Quigley CA (September 2004). "Ascertainment bias in Turner syndrome: new insights from girls who were diagnosed incidentally in prenatal life". Pediatrics. 114 (3): 640–644. doi:10.1542/peds.2003-1122-L. PMID15342833. S2CID22596252.
1 2 3 Oliveira CS, Alves C (October 2011). "The role of the SHOX gene in the pathophysiology of Turner syndrome". Endocrinologia y Nutricion. 58 (8): 433–442. doi:10.1016/j.endoen.2011.06.003. PMID21925981.
↑ Kansra AR, Donohoue PA (2011). "Hypofunction of the Ovaries". In Kliegman R (ed.). Nelson Textbook of Pediatrics (19thed.). Amsterdam: Elsevier. p.7093. ISBN978-1-4377-0755-7.
↑ Percy M, Thompson MD, Brown I, Fung WA (2016). "Other Syndromes and Conditions Associated with Intellectual and Developmental Disabilities". In Wehmeyer ML, Brown I, Percy M, Fung WA, Shogren KA (eds.). A Comprehensive Guide to Intellectual and Developmental Disabilities (2nded.). Baltimore, Maryland: Brookes Publishing. p.297. ISBN978-1-59857-602-3.
1 2 3 Lowenstein EJ, Kim KH, Glick SA (May 2004). "Turner's syndrome in dermatology". Journal of the American Academy of Dermatology. 50 (5): 767–776. doi:10.1016/j.jaad.2003.07.031. PMID15097963.
↑ Reed T (1981). "Dermatoglyphics in medicine--problems and use in suspected chromosome abnormalities". American Journal of Medical Genetics. 8 (4): 411–429. doi:10.1002/ajmg.1320080407. PMID7018239.
↑ Turtle EJ, Sule AA, Webb DJ, Bath LE (July 2015). "Aortic dissection in children and adolescents with Turner syndrome: risk factors and management recommendations". Archives of Disease in Childhood. 100 (7): 662–666. doi:10.1136/archdischild-2014-307080. PMID25573747. S2CID206857477.
↑ Fanos V, Schena S, Dal Moro A, Portuese A, Antoniazzi F (August 2000). "Multicystic kidney dysplasia and Turner syndrome: two cases and a literature review". Pediatric Nephrology. 14 (8–9): 754–757. doi:10.1007/PL00013430. PMID10955920. S2CID42881441.
1 2 3 Chen J, Guo M, Luo M, Deng S, Tian Q (August 2021). "Clinical characteristics and management of Turner patients with a small supernumerary marker chromosome". Gynecological Endocrinology. 37 (8): 730–734. doi:10.1080/09513590.2021.1911992. PMID33870841. S2CID233298107.
↑ Bonnard Å, Bark R, Hederstierna C (March 2019). "Clinical update on sensorineural hearing loss in Turner syndrome and the X-chromosome". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 181 (1): 18–24. doi:10.1002/ajmg.c.31673. PMID30632288. S2CID58589784.
↑ Practice Committee of the American Society for Reproductive Medicine (February 2012). "Increased maternal cardiovascular mortality associated with pregnancy in women with Turner syndrome". Fertility and Sterility. 97 (2): 282–284. doi:10.1016/j.fertnstert.2011.11.049 (inactive 1 November 2024). PMID22192347.{{cite journal}}: CS1 maint: DOI inactive as of November 2024 (link)
↑ Rovet JF (2019). "The Cognitive and Neuropsychological Characteristics of Females with Turner Syndrome". Sex Chromosome Abnormalities and Human Behavior. pp.38–77. doi:10.1201/9780429305870-3. ISBN978-0-429-30587-0.
↑ McCauley E, Feuillan P, Kushner H, Ross JL (December 2001). "Psychosocial development in adolescents with Turner syndrome". Journal of Developmental and Behavioral Pediatrics. 22 (6): 360–365. doi:10.1097/00004703-200112000-00003. PMID11773800. S2CID39749059.
↑ Christopoulos P, Deligeoroglou E, Laggari V, Christogiorgos S, Creatsas G (March 2008). "Psychological and behavioural aspects of patients with Turner syndrome from childhood to adulthood: a review of the clinical literature". Journal of Psychosomatic Obstetrics and Gynaecology. 29 (1): 45–51. doi:10.1080/01674820701577078. PMID17852655. S2CID8149629.
↑ Monroy N, López M, Cervantes A, García-Cruz D, Zafra G, Canún S, etal. (January 2002). "Microsatellite analysis in Turner syndrome: parental origin of X chromosomes and possible mechanism of formation of abnormal chromosomes". American Journal of Medical Genetics. 107 (3): 181–189. doi:10.1002/ajmg.10113. PMID11807897.
1 2 Uematsu A, Yorifuji T, Muroi J, Kawai M, Mamada M, Kaji M, etal. (August 2002). "Parental origin of normal X chromosomes in Turner syndrome patients with various karyotypes: implications for the mechanism leading to generation of a 45,X karyotype". American Journal of Medical Genetics. 111 (2): 134–139. doi:10.1002/ajmg.10506. PMID12210339.
1 2 Ford CE, Jones KW, Polani PE, De Almeida JC, Briggs JH (April 1959). "A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner's syndrome)". Lancet. 273 (7075): 711–713. doi:10.1016/S0140-6736(59)91893-8. PMID13642858.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.