
A premature ventricular contraction (PVC) is a common event where the heartbeat is initiated by Purkinje fibers in the ventricles rather than by the sinoatrial node. PVCs may cause no symptoms or may be perceived as a "skipped beat" or felt as palpitations in the chest. PVCs do not usually pose any danger.

Propafenone, sold under the brand name Rythmol among others, is a class 1c anti-arrhythmic medication, which is used to treat illnesses associated with rapid heart beat such as atrial and ventricular arrhythmias.
Antiarrhythmic agents, also known as cardiac dysrhythmia medications, are a class of drugs that are used to suppress abnormally fast rhythms (tachycardias), such as atrial fibrillation, supraventricular tachycardia and ventricular tachycardia.

Quinidine is a class IA antiarrhythmic agent used to treat heart rhythm disturbances. It is a diastereomer of antimalarial agent quinine, originally derived from the bark of the cinchona tree. The drug causes increased action potential duration, as well as a prolonged QT interval. As of 2019, its IV formulation is no longer being manufactured for use in the United States.

Amiodarone is an antiarrhythmic medication used to treat and prevent a number of types of cardiac dysrhythmias. This includes ventricular tachycardia (VT), ventricular fibrillation (VF), and wide complex tachycardia, as well as atrial fibrillation and paroxysmal supraventricular tachycardia. Evidence in cardiac arrest, however, is poor. It can be given by mouth, intravenously, or intraosseously. When used by mouth, it can take a few weeks for effects to begin.

Torsades de pointes, torsade de pointes or torsades des pointes is a specific type of abnormal heart rhythm that can lead to sudden cardiac death. It is a polymorphic ventricular tachycardia that exhibits distinct characteristics on the electrocardiogram (ECG). It was described by French physician François Dessertenne in 1966. Prolongation of the QT interval can increase a person's risk of developing this abnormal heart rhythm, occurring in between 1% and 10% of patients who receive QT-prolonging antiarrhythmic drugs.

Sotalol, sold under the brand name Betapace among others, is a medication used to treat and prevent abnormal heart rhythms. Evidence does not support a decreased risk of death with long term use. It is taken by mouth or given by injection into a vein.

Procainamide (PCA) is a medication of the antiarrhythmic class used for the treatment of cardiac arrhythmias. It is a sodium channel blocker of cardiomyocytes; thus it is classified by the Vaughan Williams classification system as class Ia. In addition to blocking the INa current, it inhibits the IKr rectifier K+ current. Procainamide is also known to induce a voltage-dependent open channel block on the batrachotoxin (BTX)-activated sodium channels in cardiomyocytes.

Disopyramide is an antiarrhythmic medication used in the treatment of ventricular tachycardia. It is a sodium channel blocker and is classified as a Class 1a anti-arrhythmic agent. Disopyramide has a negative inotropic effect on the ventricular myocardium, significantly decreasing the contractility. Disopyramide also has an anticholinergic effect on the heart which accounts for many adverse side effects. Disopyramide is available in both oral and intravenous forms, and has a low degree of toxicity.

Acecainide is an antiarrhythmic drug. Chemically, it is the N-acetylated metabolite of procainamide. It is a Class III antiarrhythmic agent, whereas procainamide is a Class Ia antiarrhythmic drug. It is only partially as active as procainamide; when checking levels, both must be included in the final calculation.
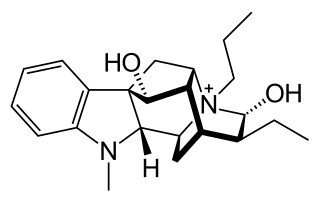
Prajmaline (Neo-gilurythmal) is a class Ia antiarrhythmic agent which has been available since the 1970s. Class Ia drugs increase the time one action potential lasts in the heart. Prajmaline is a semi-synthetic propyl derivative of ajmaline, with a higher bioavailability than its predecessor. It acts to stop arrhythmias of the heart through a frequency-dependent block of cardiac sodium channels.
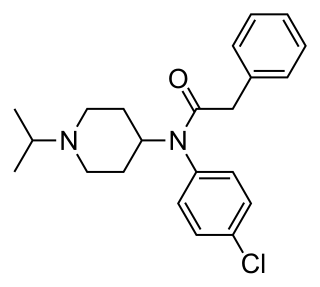
Lorcainide is a Class 1c antiarrhythmic agent that is used to help restore normal heart rhythm and conduction in patients with premature ventricular contractions, ventricular tachycardiac and Wolff–Parkinson–White syndrome. Lorcainide was developed by Janssen Pharmaceutica (Belgium) in 1968 under the commercial name Remivox and is designated by code numbers R-15889 or Ro 13-1042/001. It has a half-life of 8.9 +- 2.3 hrs which may be prolonged to 66 hrs in people with cardiac disease.

Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited genetic disorder that predisposes those affected to potentially life-threatening abnormal heart rhythms or arrhythmias. The arrhythmias seen in CPVT typically occur during exercise or at times of emotional stress, and classically take the form of bidirectional ventricular tachycardia or ventricular fibrillation. Those affected may be asymptomatic, but they may also experience blackouts or even sudden cardiac death.
Afterdepolarizations are abnormal depolarizations of cardiac myocytes that interrupt phase 2, phase 3, or phase 4 of the cardiac action potential in the electrical conduction system of the heart. Afterdepolarizations may lead to cardiac arrhythmias. Afterdepolarization is commonly a consequence of myocardial infarction, cardiac hypertrophy, or heart failure. It may also result from congenital mutations associated with calcium channels and sequestration.

Pilsicainide (INN) is an antiarrhythmic agent. It is marketed in Japan as サンリズム (Sunrythm). It was developed by Suntory Holdings Limited and first released in 1991. The JAN applies to the hydrochloride salt, pilsicainide hydrochloride.
Sodium channel blockers are drugs which impair the conduction of sodium ions (Na+) through sodium channels.

Moracizine or moricizine, sold under the trade name Ethmozine, is an antiarrhythmic of class IC. It was used for the prophylaxis and treatment of serious and life-threatening ventricular arrhythmias, but was withdrawn in 2007 for commercial reasons.

Arrhythmias, also known as cardiac arrhythmias, heart arrhythmias, or dysrhythmias, are irregularities in the heartbeat, including when it is too fast or too slow. A resting heart rate that is too fast – above 100 beats per minute in adults – is called tachycardia, and a resting heart rate that is too slow – below 60 beats per minute – is called bradycardia. Some types of arrhythmias have no symptoms. Symptoms, when present, may include palpitations or feeling a pause between heartbeats. In more serious cases, there may be lightheadedness, passing out, shortness of breath, chest pain, or decreased level of consciousness. While most cases of arrhythmia are not serious, some predispose a person to complications such as stroke or heart failure. Others may result in sudden death.
The Cardiac Arrhythmia Suppression Trial (CAST) was a double-blind, randomized, controlled study designed to test the hypothesis that suppression of premature ventricular complexes (PVC) with class I antiarrhythmic agents after a myocardial infarction (MI) would reduce mortality. It was conducted between 1986 and 1989 and included over 1700 patients in 27 centres. The study found that the tested drugs increased mortality instead of lowering it as was expected. The publication of these results in 1991/92, in combination with large follow-up studies for drugs that had not been tested in CAST, led to a paradigm shift in the treatment of MI patients. Class I and III antiarrhythmics are now only used with extreme caution after MI, or they are contraindicated completely. Heart Rhythm Society Distinguished Scientist D. George Wyse was a member of the CAST trial's steering and executive committees.
QT prolongation is a measure of delayed ventricular repolarisation, which means the heart muscle takes longer than normal to recharge between beats. It is an electrical disturbance which can be seen on an electrocardiogram (ECG). Excessive QT prolongation can trigger tachycardias such as torsades de pointes (TdP). QT prolongation is an established side effect of antiarrhythmics, but can also be caused by a wide range of non-cardiac medicines, including antibiotics, antidepressants, antihistamines, opioids, and complementary medicines. On an ECG, the QT interval represents the summation of action potentials in cardiac muscle cells, which can be caused by an increase in inward current through sodium or calcium channels, or a decrease in outward current through potassium channels. By binding to and inhibiting the “rapid” delayed rectifier potassium current protein, certain drugs are able to decrease the outward flow of potassium ions and extend the length of phase 3 myocardial repolarization, resulting in QT prolongation.

