




Alkaline phosphatase, tissue-nonspecific isozyme is an enzyme that in humans is encoded by the ALPL gene. [5] [6]
Alkaline phosphatase, tissue-nonspecific isozyme is an enzyme that in humans is encoded by the ALPL gene. [5] [6]
There are at least four distinct but related alkaline phosphatases: intestinal, placental, placental-like, and liver/bone/kidney (tissue-nonspecific). The first three are located together on chromosome 2, whereas the tissue-nonspecific form is located on chromosome 1. The product of this gene is a membrane-bound glycosylated enzyme that is expressed in a variety of tissues and is, therefore, referred to as the tissue-nonspecific form of the enzyme. A proposed function of this form of the enzyme is in regulating matrix mineralization through its ability to degrade mineralization-inhibiting pyrophosphate. Mice that lack a functional form of this enzyme (gene knockout mice) show abnormal skeletal and dental development including a mineralization deficiency called osteomalacia/odontomalacia (hypomineralization of bones and teeth). [7] [8] [9] [10] Humans with inactivating mutations in the ALPL gene likewise have variable degrees of mineralization defects depending on the location of the mutation in the ALPL gene. [11] [12]
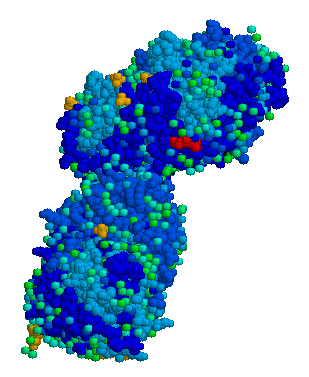
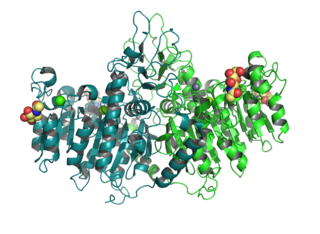
Tissue Non-Specific Alkaline Phosphatase (TNAP), encoded by the ALPL gene, exhibits an intriguing octameric structure as revealed by X-ray crystallography. [13] This distinct arrangement consists of four individual dimeric TNAP units. Structural studies on homologs of TNAP, namely human (ALPP) [14] and Escherichia coli (ecPhoA), [15] have identified the dimer as the minimal stable unit of TNAP. Notably, a single TNAP protein contains four metal ion binding sites: two Zn2+ sites and one Mg2+ site situated in the reaction center, and one Ca2+ site within the regulatory pocket. The octameric state observed in TNAP is unique compared to previously characterized alkaline phosphatases, all of which have been found in a dimeric state.
This enzyme has been linked directly to a disorder known as hypophosphatasia, a disorder that is characterized by low serum ALP and undermineralised bone (osteomalacia). The character of this disorder can vary, however, depending on the specific mutation, since this determines age of onset and severity of symptoms.
The severity of symptoms ranges from premature loss of deciduous teeth with no bone abnormalities to stillbirth [16] depending upon which amino acid [17] [18] is changed in the ALPL gene. Mutations in the ALPL gene lead to varying low activity of the enzyme tissue-nonspecific alkaline phosphatase (TNSALP) resulting in hypophosphatasia (HPP). [19] There are different clinical forms of HPP which can be inherited by an autosomal recessive trait or autosomal dominant trait, [16] the former causing more severe forms of the disease. Alkaline phosphatase allows for mineralization of calcium and phosphorus by bones and teeth. [19] ALPL gene mutation leads to insufficient TNSALP enzyme and allows for an accumulation of chemicals such as inorganic pyrophosphate [19] to indirectly cause elevated calcium levels in the body and lack of bone calcification.
The mutation E174K, where a glycine is converted to an alanine amino acid at the 571st position of its respective polypeptide chain, is a result of an ancestral mutation that occurred in Caucasians and shows a mild form of HPP. [16]
Wilson's disease is a genetic disorder in which excess copper builds up in the body. Symptoms are typically related to the brain and liver. Liver-related symptoms include vomiting, weakness, fluid build-up in the abdomen, swelling of the legs, yellowish skin, and itchiness. Brain-related symptoms include tremors, muscle stiffness, trouble in speaking, personality changes, anxiety, and psychosis.
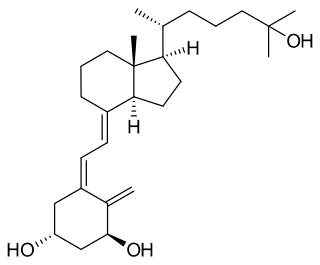
Osteomalacia is a disease characterized by the softening of the bones caused by impaired bone metabolism primarily due to inadequate levels of available phosphate, calcium, and vitamin D, or because of resorption of calcium. The impairment of bone metabolism causes inadequate bone mineralization. Osteomalacia in children is known as rickets, and because of this, use of the term "osteomalacia" is often restricted to the milder, adult form of the disease. Signs and symptoms can include diffuse body pains, muscle weakness, and fragility of the bones. In addition to low systemic levels of circulating mineral ions that result in decreased bone and tooth mineralization, accumulation of mineralization-inhibiting proteins and peptides, and small inhibitory molecules, can occur in the extracellular matrix of bones and teeth, contributing locally to cause matrix hypomineralization (osteomalacia/odontomalacia). A relationship describing local, physiologic double-negative regulation of mineralization has been termed the Stenciling Principle of mineralization, whereby enzyme-substrate pairs imprint mineralization patterns into the extracellular matrix by degrading mineralization inhibitors. The Stenciling Principle for mineralization is particularly relevant to the osteomalacia and odontomalacia observed in hypophosphatasia (HPP) and X-linked hypophosphatemia (XLH).
Gaucher's disease or Gaucher disease (GD) is a genetic disorder in which glucocerebroside accumulates in cells and certain organs. The disorder is characterized by bruising, fatigue, anemia, low blood platelet count and enlargement of the liver and spleen, and is caused by a hereditary deficiency of the enzyme glucocerebrosidase, which acts on glucocerebroside. When the enzyme is defective, glucocerebroside accumulates, particularly in white blood cells and especially in macrophages. Glucocerebroside can collect in the spleen, liver, kidneys, lungs, brain, and bone marrow.

The enzyme alkaline phosphatase is a phosphatase with the physiological role of dephosphorylating compounds. The enzyme is found across a multitude of organisms, prokaryotes and eukaryotes alike, with the same general function, but in different structural forms suitable to the environment they function in. Alkaline phosphatase is found in the periplasmic space of E. coli bacteria. This enzyme is heat stable and has its maximum activity at high pH. In humans, it is found in many forms depending on its origin within the body – it plays an integral role in metabolism within the liver and development within the skeleton. Due to its widespread prevalence in these areas, its concentration in the bloodstream is used by diagnosticians as a biomarker in helping determine diagnoses such as hepatitis or osteomalacia.
Hypophosphatasia (; also called deficiency of alkaline phosphatase, phosphoethanolaminuria, or Rathbun's syndrome; sometimes abbreviated HPP) is a rare, and sometimes fatal, inherited metabolic bone disease. Clinical symptoms are heterogeneous, ranging from the rapidly fatal, perinatal variant, with profound skeletal hypomineralization, respiratory compromise or vitamin B6 dependent seizures to a milder, progressive osteomalacia later in life. Tissue non-specific alkaline phosphatase (TNSALP) deficiency in osteoblasts and chondrocytes impairs bone mineralization, leading to rickets or osteomalacia. The pathognomonic finding is subnormal serum activity of the TNSALP enzyme, which is caused by one of 388 genetic mutations identified to date, in the gene encoding TNSALP. Genetic inheritance is autosomal recessive for the perinatal and infantile forms but either autosomal recessive or autosomal dominant in the milder forms.
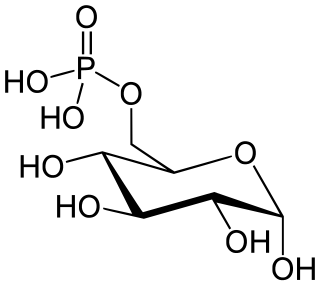
The enzyme glucose 6-phosphatase (EC 3.1.3.9, G6Pase; systematic name D-glucose-6-phosphate phosphohydrolase) catalyzes the hydrolysis of glucose 6-phosphate, resulting in the creation of a phosphate group and free glucose:
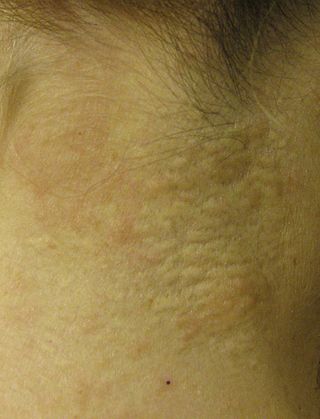
Pseudoxanthoma elasticum (PXE) is a genetic disease that causes mineralization of elastic fibers in some tissues. The most common problems arise in the skin and eyes, and later in blood vessels in the form of premature atherosclerosis. PXE is caused by autosomal recessive mutations in the ABCC6 gene on the short arm of chromosome 16 (16p13.1).
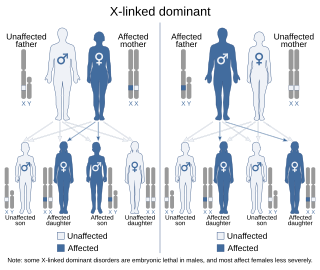
X-linked hypophosphatemia (XLH) is an X-linked dominant form of rickets that differs from most cases of dietary deficiency rickets in that vitamin D supplementation does not cure it. It can cause bone deformity including short stature and genu varum (bow-leggedness). It is associated with a mutation in the PHEX gene sequence (Xp.22) and subsequent inactivity of the PHEX protein. PHEX mutations lead to an elevated circulating (systemic) level of the hormone FGF23 which results in renal phosphate wasting, and local elevations of the mineralization/calcification-inhibiting protein osteopontin in the extracellular matrix of bones and teeth. An inactivating mutation in the PHEX gene results in an increase in systemic circulating FGF23, and a decrease in the enzymatic activity of the PHEX enzyme which normally removes (degrades) mineralization-inhibiting osteopontin protein; in XLH, the decreased PHEX enzyme activity leads to an accumulation of inhibitory osteopontin locally in bones and teeth to block mineralization which, along with renal phosphate wasting, both cause osteomalacia and odontomalacia.

CYP27A1 is a gene encoding a cytochrome P450 oxidase, and is commonly known as sterol 27-hydroxylase. This enzyme is located in many different tissues where it is found within the mitochondria. It is most prominently involved in the biosynthesis of bile acids.
Phosphate-regulating endopeptidase homolog X-linked also known as phosphate-regulating gene with homologies to endopeptidases on the X chromosome or metalloendopeptidase homolog PEX is an enzyme that in humans is encoded by the PHEX gene. This gene contains 18 exons and is located on the X chromosome.
N-acetylgalactosamine-6-sulfatase is an enzyme that, in humans, is encoded by the GALNS gene.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

HNF1 homeobox B, also known as HNF1B or transcription factor 2 (TCF2), is a human gene.

3-oxoacid CoA-transferase 1 (OXCT1) is an enzyme that in humans is encoded by the OXCT1 gene. It is also known as succinyl-CoA-3-oxaloacid CoA transferase (SCOT). Mutations in the OXCT1 gene are associated with succinyl-CoA:3-oxoacid CoA transferase deficiency. This gene encodes a member of the 3-oxoacid CoA-transferase gene family. The encoded protein is a homodimeric mitochondrial matrix enzyme that plays a central role in extrahepatic ketone body catabolism by catalyzing the reversible transfer of coenzyme A (CoA) from succinyl-CoA to acetoacetate.
Alkaline phosphatase, placental type also known as placental alkaline phosphatase (PLAP) is an allosteric enzyme that in humans is encoded by the ALPP gene.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.

Elevated alkaline phosphatase occurs when levels of alkaline phosphatase (ALP) exceed the reference range. This group of enzymes has a low substrate specificity and catalyzes the hydrolysis of phosphate esters in a basic environment. The major function of alkaline phosphatase is transporting chemicals across cell membranes. Alkaline phosphatases are present in many human tissues, including bone, intestine, kidney, liver, placenta and white blood cells. Damage to these tissues causes the release of ALP into the bloodstream. Elevated levels can be detected through a blood test. Elevated alkaline phosphate is associated with certain medical conditions or syndromes. It serves as a significant indicator for certain medical conditions, diseases and syndromes.
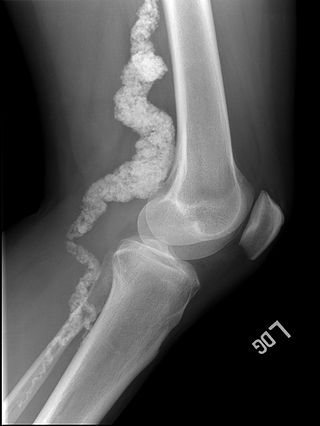
Arterial calcification due to deficiency of CD73 (ACDC) is a rare genetic disorder that causes calcium buildup in the arteries and joints of the hands and feet, and other areas below the waist. Although patients exhibiting these symptoms have been identified as early as 1914, this disorder had not been studied extensively until recently. The identification of the specific ACDC gene and mutations occurred in 2011. ACDC is caused by a mutation in the NT5E gene, which prevents calcium-removing agents from functioning,. Patients with this mutation experience chronic pain, difficulty moving, and increased risk of cardiovascular problems. In experiments at the molecular level, treatment with adenosine or a phosphatase inhibitor reversed and prevented calcification, suggesting they could be used as possible treatment methods. There is currently no cure for ACDC, and patients have limited treatment options which focus primarily on removal of blood calcium and improving mobility.
Alkaline phosphatase, placental-like 2 is a protein that in humans is encoded by the ALPPL2 gene.
Asfotase alfa, sold under the brand name Strensiq, is a medication used in the treatment of people with perinatal/infantile- and juvenile-onset hypophosphatasia.