An intron is any nucleotide sequence within a gene that is not expressed or operative in the final RNA product. The word intron is derived from the term intragenic region, i.e., a region inside a gene. The term intron refers to both the DNA sequence within a gene and the corresponding RNA sequence in RNA transcripts. The non-intron sequences that become joined by this RNA processing to form the mature RNA are called exons.

Protein biosynthesis is a core biological process, occurring inside cells, balancing the loss of cellular proteins through the production of new proteins. Proteins perform a number of critical functions as enzymes, structural proteins or hormones. Protein synthesis is a very similar process for both prokaryotes and eukaryotes but there are some distinct differences.

RNA splicing is a process in molecular biology where a newly-made precursor messenger RNA (pre-mRNA) transcript is transformed into a mature messenger RNA (mRNA). It works by removing all the introns and splicing back together exons. For nuclear-encoded genes, splicing occurs in the nucleus either during or immediately after transcription. For those eukaryotic genes that contain introns, splicing is usually needed to create an mRNA molecule that can be translated into protein. For many eukaryotic introns, splicing occurs in a series of reactions which are catalyzed by the spliceosome, a complex of small nuclear ribonucleoproteins (snRNPs). There exist self-splicing introns, that is, ribozymes that can catalyze their own excision from their parent RNA molecule. The process of transcription, splicing and translation is called gene expression, the central dogma of molecular biology.
Gene expression is the process by which information from a gene is used in the synthesis of a functional gene product that enables it to produce end products, proteins or non-coding RNA, and ultimately affect a phenotype. These products are often proteins, but in non-protein-coding genes such as transfer RNA (tRNA) and small nuclear RNA (snRNA), the product is a functional non-coding RNA. The process of gene expression is used by all known life—eukaryotes, prokaryotes, and utilized by viruses—to generate the macromolecular machinery for life.
The coding region of a gene, also known as the coding sequence (CDS), is the portion of a gene's DNA or RNA that codes for a protein. Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes. This can further assist in mapping the human genome and developing gene therapy.

Alternative splicing, or alternative RNA splicing, or differential splicing, is an alternative splicing process during gene expression that allows a single gene to produce different splice variants. For example, some exons of a gene may be included within or excluded from the final RNA product of the gene. This means the exons are joined in different combinations, leading to different splice variants. In the case of protein-coding genes, the proteins translated from these splice variants may contain differences in their amino acid sequence and in their biological functions.
A protein isoform, or "protein variant", is a member of a set of highly similar proteins that originate from a single gene or gene family and are the result of genetic differences. While many perform the same or similar biological roles, some isoforms have unique functions. A set of protein isoforms may be formed from alternative splicings, variable promoter usage, or other post-transcriptional modifications of a single gene; post-translational modifications are generally not considered. Through RNA splicing mechanisms, mRNA has the ability to select different protein-coding segments (exons) of a gene, or even different parts of exons from RNA to form different mRNA sequences. Each unique sequence produces a specific form of a protein.

SR proteins are a conserved family of proteins involved in RNA splicing. SR proteins are named because they contain a protein domain with long repeats of serine and arginine amino acid residues, whose standard abbreviations are "S" and "R" respectively. SR proteins are ~200-600 amino acids in length and composed of two domains, the RNA recognition motif (RRM) region and the RS domain. SR proteins are more commonly found in the nucleus than the cytoplasm, but several SR proteins are known to shuttle between the nucleus and the cytoplasm.

Familial dysautonomia (FD), also known as Riley-Day syndrome, is a rare, progressive, recessive genetic disorder of the autonomic nervous system that affects the development and survival of sensory, sympathetic, and some parasympathetic neurons in the autonomic and sensory nervous system.
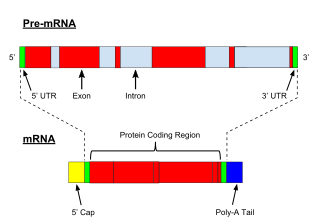
A primary transcript is the single-stranded ribonucleic acid (RNA) product synthesized by transcription of DNA, and processed to yield various mature RNA products such as mRNAs, tRNAs, and rRNAs. The primary transcripts designated to be mRNAs are modified in preparation for translation. For example, a precursor mRNA (pre-mRNA) is a type of primary transcript that becomes a messenger RNA (mRNA) after processing.
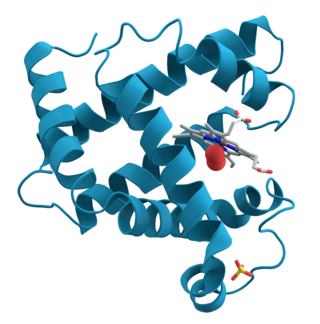
L1, also known as L1CAM, is a transmembrane protein member of the L1 protein family, encoded by the L1CAM gene. This protein, of 200-220 kDa, is a neuronal cell adhesion molecule with a strong implication in cell migration, adhesion, neurite outgrowth, myelination and neuronal differentiation. It also plays a key role in treatment-resistant cancers due to its function. It was first identified in 1984 by M. Schachner who found the protein in post-mitotic mice neurons.
Transcriptional modification or co-transcriptional modification is a set of biological processes common to most eukaryotic cells by which an RNA primary transcript is chemically altered following transcription from a gene to produce a mature, functional RNA molecule that can then leave the nucleus and perform any of a variety of different functions in the cell. There are many types of post-transcriptional modifications achieved through a diverse class of molecular mechanisms.

Neurofibromin 1 (NF1) is a gene in humans that is located on chromosome 17. NF1 codes for neurofibromin, a GTPase-activating protein that negatively regulates RAS/MAPK pathway activity by accelerating the hydrolysis of Ras-bound GTP. NF1 has a high mutation rate and mutations in NF1 can alter cellular growth control, and neural development, resulting in neurofibromatosis type 1. Symptoms of NF1 include disfiguring cutaneous neurofibromas (CNF), café au lait pigment spots, plexiform neurofibromas (PN), skeletal defects, optic nerve gliomas, life-threatening malignant peripheral nerve sheath tumors (MPNST), pheochromocytoma, attention deficits, learning deficits and other cognitive disabilities.

NF-kappa-B essential modulator (NEMO) also known as inhibitor of nuclear factor kappa-B kinase subunit gamma (IKK-γ) is a protein that in humans is encoded by the IKBKG gene. NEMO is a subunit of the IκB kinase complex that activates NF-κB. The human gene for IKBKG is located on the chromosome band Xq28. Multiple transcript variants encoding different isoforms have been found for this gene.
Eukaryotic transcription is the elaborate process that eukaryotic cells use to copy genetic information stored in DNA into units of transportable complementary RNA replica. Gene transcription occurs in both eukaryotic and prokaryotic cells. Unlike prokaryotic RNA polymerase that initiates the transcription of all different types of RNA, RNA polymerase in eukaryotes comes in three variations, each translating a different type of gene. A eukaryotic cell has a nucleus that separates the processes of transcription and translation. Eukaryotic transcription occurs within the nucleus where DNA is packaged into nucleosomes and higher order chromatin structures. The complexity of the eukaryotic genome necessitates a great variety and complexity of gene expression control.

Actin-like protein 7A is a protein that in humans is encoded by the ACTL7A gene.
RNA polymerase II holoenzyme is a form of eukaryotic RNA polymerase II that is recruited to the promoters of protein-coding genes in living cells. It consists of RNA polymerase II, a subset of general transcription factors, and regulatory proteins known as SRB proteins.
Hereditary sensory and autonomic neuropathy (HSAN) or hereditary sensory neuropathy (HSN) is a condition used to describe any of the types of this disease which inhibit sensation.
Numerous key discoveries in biology have emerged from studies of RNA, including seminal work in the fields of biochemistry, genetics, microbiology, molecular biology, molecular evolution, and structural biology. As of 2010, 30 scientists have been awarded Nobel Prizes for experimental work that includes studies of RNA. Specific discoveries of high biological significance are discussed in this article.
The TREX (TRanscription-EXport) complex is a conserved eukaryotic multi-protein complex that couples mRNA transcription and nuclear export. The TREX complex travels across transcribed genes with RNA polymerase II. TREX binds mRNA and recruits transport proteins NXF1 and NXT1, which shuttle the mRNA out of the nucleus. The TREX complex plays an important role in genome stability and neurodegenerative diseases.
