Related Research Articles

Osteopetrosis, literally "stone bone", also known as marble bone disease or Albers-Schönberg disease, is an extremely rare inherited disorder whereby the bones harden, becoming denser, in contrast to more prevalent conditions like osteoporosis, in which the bones become less dense and more brittle, or osteomalacia, in which the bones soften. Osteopetrosis can cause bones to dissolve and break.

Tuberous sclerosis complex (TSC) is a rare multisystem autosomal dominant genetic disease that causes non-cancerous tumours to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, lung disease, and kidney disease.
Gardner's syndrome is a subtype of familial adenomatous polyposis (FAP). Gardner syndrome is an autosomal dominant form of polyposis characterized by the presence of multiple polyps in the colon together with tumors outside the colon. The extracolonic tumors may include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas, as well as the occurrence of desmoid tumors in approximately 15% of affected individuals.

The term fibromatosis refers to a group of soft tissue tumors which have certain characteristics in common, including absence of cytologic and clinical malignant features, a histology consistent with proliferation of well-differentiated fibroblasts, an infiltrative growth pattern, and aggressive clinical behavior with frequent local recurrence. It is classed by the World Health Organization as an intermediate soft tissue tumor related to the sarcoma family. Arthur Purdy Stout coined the term fibromatosis, in 1954.

A benign tumor is a mass of cells (tumor) that does not invade neighboring tissue or metastasize. Compared to malignant (cancerous) tumors, benign tumors generally have a slower growth rate. Benign tumors have relatively well differentiated cells. They are often surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.
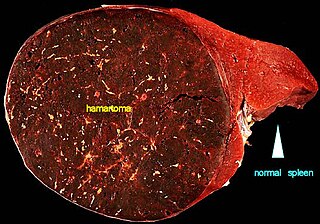
A hamartoma is a mostly benign, local malformation of cells that resembles a neoplasm of local tissue but is usually due to an overgrowth of multiple aberrant cells, with a basis in a systemic genetic condition, rather than a growth descended from a single mutated cell (monoclonality), as would typically define a benign neoplasm/tumor. Despite this, many hamartomas are found to have clonal chromosomal aberrations that are acquired through somatic mutations, and on this basis the term hamartoma is sometimes considered synonymous with neoplasm. Hamartomas are by definition benign, slow-growing or self-limiting, though the underlying condition may still predispose the individual towards malignancies.

Multiple endocrine neoplasia type 1 (MEN-1) is one of a group of disorders, the multiple endocrine neoplasias, that affect the endocrine system through development of neoplastic lesions in pituitary, parathyroid gland and pancreas. Individuals suffering from this disorder are prone to developing multiple endocrine and nonendocrine tumors. It was first described by Paul Wermer in 1954.

Cowden syndrome is an autosomal dominant inherited condition characterized by benign overgrowths called hamartomas as well as an increased lifetime risk of breast, thyroid, uterine, and other cancers. It is often underdiagnosed due to variability in disease presentation, but 99% of patients report mucocutaneous symptoms by age 20–29. Despite some considering it a primarily dermatologic condition, Cowden's syndrome is a multi-system disorder that also includes neurodevelopmental disorders such as macrocephaly.
Phakomatoses, also known neurocutaneous syndromes, are a group of multisystemic diseases that most prominently affect structures primarily derived from the ectoderm such as the central nervous system, skin and eyes. The majority of phakomatoses are single-gene disorders that may be inherited in an autosomal dominant, autosomal recessive or X-linked pattern. Presentations may vary dramatically between patients with the same particular syndrome due to mosaicism, variable expressivity, and penetrance.

Schwannomatosis is an extremely rare genetic disorder closely related to the more-common disorder neurofibromatosis (NF). Originally described in Japanese patients, it consists of multiple cutaneous schwannomas, central nervous system tumors, and other neurological complications, excluding hallmark signs of NF. The exact frequency of schwannomatosis cases is unknown, although some populations have noted frequencies as few as 1 case per 1.7 million people.

Aggressive fibromatosis or desmoid tumor is a rare condition. Desmoid tumors are a type of fibromatosis and related to sarcoma, though without the ability to spread throughout the body (metastasize). The tumors arise from cells called fibroblasts, which are found throughout the body and provide structural support, protection to the vital organs, and play a critical role in wound healing. These tumors tend to occur in women in their thirties, but can occur in anyone at any age. They can be either relatively slow-growing or malignant. However, aggressive fibromatosis is locally aggressive and can cause life-threatening problems or even death when the tumors compress vital organs such as intestines, kidneys, lungs, blood vessels, or nerves. The condition is rarely fatal. Most cases are sporadic, but some are associated with familial adenomatous polyposis (FAP). Approximately 10% of individuals with Gardner's syndrome, a type of FAP with extracolonic features, have desmoid tumors.

Granular cell tumor is a tumor that can develop on any skin or mucosal surface, but occurs on the tongue 40% of the time.
Infantile Refsum disease (IRD) is a rare autosomal recessive congenital peroxisomal biogenesis disorder within the Zellweger spectrum. These are disorders of the peroxisomes that are clinically similar to Zellweger syndrome and associated with mutations in the PEX family of genes. IRD is associated with deficient phytanic acid catabolism, as is adult Refsum disease, but they are different disorders that should not be confused.
Infantile digital fibromatosis (IDF), also termed inclusion body fibromatosis, Reye tumor, or Reye's tumor, usually occurs as a single, small, asymptomatic, nodule in the dermis on a finger or toe of infants and young children. IMF is a rare disorder with approximately 200 cases reported in the medical literature as of 2021. The World Health Organization in 2020 classified these nodules as a specific benign tumor type in the category of fibroblastic and myofibroblastic tumors. IDF was first described by the Australian pathologist, Douglas Reye, in 1965.
Infantile myofibromatosis (IMF) is a rare tumor found in 1 in 150,000 to 1 in 400,000 live births. It is nonetheless the most common tumor derived from fibrous connective tissue that occurs primarily in infants and young children. IMF tumors are benign in the sense that they do not metastasize to distant tissues although when occurring in the viscera, i.e. internal organs, carry guarded to poor prognoses and can be life-threatening, particularly in newborns and young infants. The condition was first described by Arthur Purdy Stout as congenital generalized fibromatosis – in which he coined the word fibromatosis – in 1954.

Mammary-type myofibroblastoma (MFB), also named mammary and extramammary myofibroblastoma, was first termed myofibrolastoma of the breast, or, more simply, either mammary myofibroblastoma (MMFB) or just myofibroblastoma. The change in this terminology occurred because the initial 1987 study and many subsequent studies found this tumor only in breast tissue. However, a 2001 study followed by numerous reports found tumors with the microscopic histopathology and other key features of mammary MFB in a wide range of organs and tissues. Further complicating the issue, early studies on MFB classified it as one of various types of spindle cell tumors that, except for MFB, were ill-defined. These other tumors, which have often been named interchangeably in different reports, are: myelofibroblastoma, benign spindle cell tumor, fibroma, spindle cell lipoma, myogenic stromal tumor, and solitary stromal tumor. Finally, studies suggest that spindle cell lipoma and cellular angiofibroma are variants of MFB. Here, the latter two tumors are tentatively classified as MFB variants but otherwise MFB is described as it is more strictly defined in most recent publications. The World Health Organization in 2020 classified mammary type myofibroblastoma tumors and myofibroblastoma tumors as separate tumor forms within the category of fibroblastic and myofibroblastic tumors.
Hereditary gingival fibromatosis (HGF), also known as idiopathic gingival hyperplasia, is a rare condition of gingival overgrowth. HGF is characterized as a benign, slowly progressive, nonhemorrhagic, fibrous enlargement of keratinized gingiva. It can cover teeth in various degrees, and can lead to aesthetic disfigurement. Fibrous enlargement is most common in areas of maxillary and mandibular tissues of both arches in the mouth. Phenotype and genotype frequency of HGF is 1:175,000 where males and females are equally affected but the cause is not entirely known. It mainly exists as an isolated abnormality but can also be associated with a multi-system syndrome.
Epstein–Barr virus–associated lymphoproliferative diseases are a group of disorders in which one or more types of lymphoid cells, i.e. B cells, T cells, NK cells, and histiocytic-dendritic cells, are infected with the Epstein–Barr virus (EBV). This causes the infected cells to divide excessively, and is associated with the development of various non-cancerous, pre-cancerous, and cancerous lymphoproliferative disorders (LPDs). These LPDs include the well-known disorder occurring during the initial infection with the EBV, infectious mononucleosis, and the large number of subsequent disorders that may occur thereafter. The virus is usually involved in the development and/or progression of these LPDs although in some cases it may be an "innocent" bystander, i.e. present in, but not contributing to, the disease.
Fibroblastic and myofibroblastic tumors (FMTs) develop from the mesenchymal stem cells which differentiate into fibroblasts and/or the myocytes/myoblasts that differentiate into muscle cells. FMTs are a heterogeneous group of soft tissue neoplasms. The World Health Organization (2020) defined tumors as being FMTs based on their morphology and, more importantly, newly discovered abnormalities in the expression levels of key gene products made by these tumors' neoplastic cells. Histopathologically, FMTs consist of neoplastic connective tissue cells which have differented into cells that have microscopic appearances resembling fibroblasts and/or myofibroblasts. The fibroblastic cells are characterized as spindle-shaped cells with inconspicuous nucleoli that express vimentin, an intracellular protein typically found in mesenchymal cells, and CD34, a cell surface membrane glycoprotein. Myofibroblastic cells are plumper with more abundant cytoplasm and more prominent nucleoli; they express smooth muscle marker proteins such as smooth muscle actins, desmin, and caldesmon. The World Health Organization further classified FMTs into four tumor forms based on their varying levels of aggressiveness: benign, intermediate, intermediate, and malignant.
Gardner fibroma (GF) is a benign fibroblastic tumor. GF tumors typically develop in the dermis and adjacent subcutaneous tissue lying just below the dermis. These tumors typically occur on the back, abdomen, and other superficial sites but in rare cases have been diagnoses in internal sites such as the retroperitoneum and around the large blood vessels in the upper thoracic cavity. The World Health Organization, 2020, classified Gardner fibroma as a benign tumor in the category of fibroblastic and myofibroblastic tumors.
References
- ↑ James W, Berger T, Elston D (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. ISBN 0-7216-2921-0.
- 1 2 3 4 5 6 Kameda N, Atobe T, Akima M, Fukuoka Y, Ikeno Y, Saito M (November 1978). "Diffuse infantile fibromatosis". Acta Pathologica Japonica. 28 (6): 957–962. doi:10.1111/j.1440-1827.1978.tb01284.x. PMID 735826. S2CID 33395556.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 "Infantile Myofibromatosis". NORD (National Organization for Rare Disorders). 2020. Retrieved 2022-07-28.
- ↑ Allen PW (September 1977). "The fibromatoses: a clinicopathologic classification based on 140 cases". The American Journal of Surgical Pathology. 1 (3): 255–270. doi:10.1097/00000478-197709000-00007. PMID 920873. S2CID 41764536.
- 1 2 3 "Desmoid Tumor - NCI". www.cancer.gov. 2019-02-27. Retrieved 2022-08-01.
- 1 2 3 "Infantile Myofibromatosis - NCI". www.cancer.gov. 2020-07-22. Retrieved 2022-07-28.
- ↑ Larralde M, Ferrari B, Martinez JP, Barbieri MA, Méndez JH, Casas J (December 2017). "Infantile myofibromatosis". Anais Brasileiros de Dermatologia. 92 (6): 854–857. doi:10.1590/abd1806-4841.20175001. PMC 5786406 . PMID 29364448.
- ↑ Guérit E, Arts F, Dachy G, Boulouadnine B, Demoulin JB (April 2021). "PDGF receptor mutations in human diseases". Cellular and Molecular Life Sciences. 78 (8): 3867–3881. doi:10.1007/s00018-020-03753-y. PMID 33449152. S2CID 231612187.
- 1 2 3 Wu D, Wang S, Oliveira DV, Del Gaudio F, Vanlandewijck M, Lebouvier T, et al. (January 2021). "The infantile myofibromatosis NOTCH3 L1519P mutation leads to hyperactivated ligand-independent Notch signaling and increased PDGFRB expression". Disease Models & Mechanisms. 14 (2): dmm046300. doi:10.1242/dmm.046300. PMC 7927659 . PMID 33509954.
- 1 2 3 "Infantile myofibromatosis - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Retrieved 2022-07-28.
- ↑ "Somatic Mutation vs. Germline Mutation". Cleveland Clinic. Retrieved 2022-07-29.
- ↑ "How do tumors grow and spread?". UMass Chan Medical School. MCCB. 28 August 2015. Retrieved 2022-07-26.
- 1 2 3 4 5 6 Hettmer S, Dachy G, Seitz G, Agaimy A, Duncan C, Jongmans M, et al. (October 2021). "Genetic testing and surveillance in infantile myofibromatosis: a report from the SIOPE Host Genome Working Group". Familial Cancer. 20 (4): 327–336. doi:10.1007/s10689-020-00204-2. PMC 8484085 . PMID 32888134.
- ↑ Carruthers V, Barnett S, Rees R, Arif T, Slater O, Ramanujachar R, et al. (July 2022). "Clinical utility of vinblastine therapeutic drug monitoring for the treatment of infantile myofibroma patients: A case series". Pediatric Blood & Cancer. 69 (7): e29722. doi:10.1002/pbc.29722. PMID 35441483. S2CID 248263569.
- ↑ Mashiah J, Hadj-Rabia S, Dompmartin A, Harroche A, Laloum-Grynberg E, Wolter M, et al. (August 2014). "Infantile myofibromatosis: a series of 28 cases". Journal of the American Academy of Dermatology. 71 (2): 264–270. doi:10.1016/j.jaad.2014.03.035. PMID 24894456.
- 1 2 Mynatt CJ, Feldman KA, Thompson LD (September 2011). "Orbital infantile myofibroma: a case report and clinicopathologic review of 24 cases from the literature". Head and Neck Pathology. 5 (3): 205–215. doi:10.1007/s12105-011-0260-4. PMC 3173528 . PMID 21512784.
- ↑ "What is Infantile Myofibromatosis?". News-Medical.net. 2022-07-07. Retrieved 2022-08-01.
- 1 2 3 4 Römer T, Wagner N, Braunschweig T, Meyer R, Elbracht M, Kontny U, Moser O (June 2021). "Aggressive infantile myofibromatosis with intestinal involvement". Molecular and Cellular Pediatrics. 8 (1): 7. doi: 10.1186/s40348-021-00117-9 . PMC 8208328 . PMID 34132909.
- ↑ Parikh A, Driscoll CA, Crowley H, York T, Dachy G, Demoulin JB, Hoffman SB (November 2020). "Diagnostic limitations and considerations in the imaging evaluation of advanced multicentric infantile myofibromatosis". Radiology Case Reports. 15 (11): 2440–2444. doi:10.1016/j.radcr.2020.09.029. PMC 7522587 . PMID 33014229.