Related Research Articles

The Golgi apparatus, also known as the Golgi complex, Golgi body, or simply the Golgi, is an organelle found in most eukaryotic cells. Part of the endomembrane system in the cytoplasm, it packages proteins into membrane-bound vesicles inside the cell before the vesicles are sent to their destination. It resides at the intersection of the secretory, lysosomal, and endocytic pathways. It is of particular importance in processing proteins for secretion, containing a set of glycosylation enzymes that attach various sugar monomers to proteins as the proteins move through the apparatus.
GTPases are a large family of hydrolase enzymes that bind to the nucleotide guanosine triphosphate (GTP) and hydrolyze it to guanosine diphosphate (GDP). The GTP binding and hydrolysis takes place in the highly conserved P-loop "G domain", a protein domain common to many GTPases.
The Coat Protein Complex II, or COPII, is a group of proteins that facilitate the formation of vesicles to transport proteins from the endoplasmic reticulum to the Golgi apparatus or endoplasmic-reticulum–Golgi intermediate compartment. This process is termed anterograde transport, in contrast to the retrograde transport associated with the COPI complex. COPII is assembled in two parts: first an inner layer of Sar1, Sec23, and Sec24 forms; then the inner coat is surrounded by an outer lattice of Sec13 and Sec31.

COPI is a coatomer, a protein complex that coats vesicles transporting proteins from the cis end of the Golgi complex back to the rough endoplasmic reticulum (ER), where they were originally synthesized, and between Golgi compartments. This type of transport is retrograde transport, in contrast to the anterograde transport associated with the COPII protein. The name "COPI" refers to the specific coat protein complex that initiates the budding process on the cis-Golgi membrane. The coat consists of large protein subcomplexes that are made of seven different protein subunits, namely α, β, β', γ, δ, ε and ζ.

Brefeldin A is a lactone antiviral produced by the fungus Penicillium brefeldianum. Brefeldin A inhibits protein transport from the endoplasmic reticulum to the golgi complex indirectly by preventing association of COP-I coat to the Golgi membrane. Brefeldin A was initially isolated with hopes to become an antiviral drug but is now primarily used in research to study protein transport.
TRAPP (TRAnsport Protein Particle) is a protein involved in particle transport between organelles.

ADP ribosylation factors (ARFs) are members of the ARF family of GTP-binding proteins of the Ras superfamily. ARF family proteins are ubiquitous in eukaryotic cells, and six highly conserved members of the family have been identified in mammalian cells. Although ARFs are soluble, they generally associate with membranes because of N-terminus myristoylation. They function as regulators of vesicular traffic and actin remodelling.
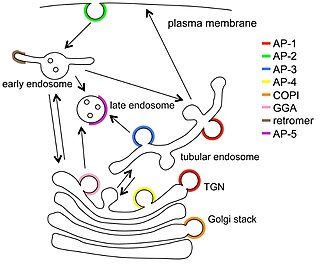
Vesicular transport adaptor proteins are proteins involved in forming complexes that function in the trafficking of molecules from one subcellular location to another. These complexes concentrate the correct cargo molecules in vesicles that bud or extrude off of one organelle and travel to another location, where the cargo is delivered. While some of the details of how these adaptor proteins achieve their trafficking specificity has been worked out, there is still much to be learned.

ADP-ribosylation factor 1 is a protein that in humans is encoded by the ARF1 gene.
Coatomer subunit beta is a protein that in humans is encoded by the COPB1 gene.
Coatomer subunit alpha is a protein that in humans is encoded by the COPA gene.

ADP-ribosylation factor GTPase-activating protein 1 is an enzyme that in humans is encoded by the ARFGAP1 gene. Two transcript variants encoding different isoforms have been found for this gene.

Coatomer subunit beta is a protein that is encoded by the COPB2 gene in humans.

Coatomer subunit gamma is a protein that in humans is encoded by the COPG gene. It is one of seven proteins in the COPI coatomer complex that coats vesicles as they bud from the Golgi complex.

ADP-ribosylation factor 4 is a protein that in humans is encoded by the ARF4 gene.

KDEL (Lys-Asp-Glu-Leu) endoplasmic reticulum protein retention receptor 1, also known as KDELR1, is a protein which in humans is encoded by the KDELR1 gene.

Chylomicron retention disease is a disorder of fat absorption. It is associated with SAR1B. Mutations in SAR1B prevent the release of chylomicrons in the circulation which leads to nutritional and developmental problems. It is a rare autosomal recessive disorder with around 40 cases reported worldwide. Since the disease allele is recessive, parents usually do not show symptoms.
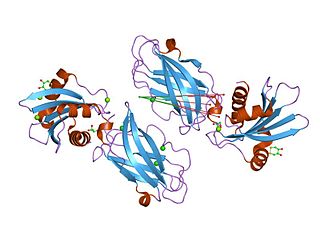
The C-terminal domain ofBeta2-adaptin is a protein domain is involved in cell trafficking by aiding import and export of substances in and out of the cell.
Exomer is a heterotetrameric protein complex similar to COPI and other adaptins. It was first described in the yeast Saccharomyces cerevisiae. Exomer is a cargo adaptor important in transporting molecules from the Golgi apparatus toward the cell membrane. The vesicles it is found on are different from COPI vesicles in that they do not appear to have a "coat" or "scaffold" around them.
Halperin-Birk syndrome (HLBKS) is a rare autosomal recessive neurodevelopmental disorder caused by a null mutation in the SEC31A gene. Signs and symptoms include intrauterine growth retardation, marked developmental delay, spastic quadriplegia with profound contractures, dysmorphism, and optic nerve atrophy with no eye fixation. Brain MRI demonstrated microcephaly and agenesis of the corpus callosum.
References
- ↑ Coatomer+Protein at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- ↑ Boehm, Markus; Bonifacino, Juan S. (October 2001). "Adaptins". Molecular Biology of the Cell. 12 (10): 2907–2920. doi:10.1091/mbc.12.10.2907. ISSN 1059-1524. PMC 60144 . PMID 11598180.
- 1 2 3 4 Gomez-Navarro, Natalia; Miller, Elizabeth A. (2016-01-25). "COP-coated vesicles". Current Biology. 26 (2): R54–R57. doi: 10.1016/j.cub.2015.12.017 . ISSN 0960-9822. PMID 26811885.
- 1 2 3 Lodish, Harvey; Berk, Arnold; Zipursky, S. Lawrence; Matsudaira, Paul; Baltimore, David; Darnell, James (2000). "Molecular Mechanisms of Vesicular Traffic". Molecular Cell Biology. 4th Edition.
- 1 2 3 Arakel, Eric C.; Schwappach, Blanche (2018-03-01). "Formation of COPI-coated vesicles at a glance". Journal of Cell Science. 131 (5): jcs209890. doi: 10.1242/jcs.209890 . hdl: 21.11116/0000-0000-F94F-0 . ISSN 0021-9533. PMID 29535154.
- 1 2 3 4 5 Duden, Rainer (2003-01-01). "ER-to-Golgi transport: COP I and COP II function (Review)". Molecular Membrane Biology. 20 (3): 197–207. doi: 10.1080/0968768031000122548 . ISSN 0968-7688. PMID 12893528. S2CID 24067181.
- 1 2 3 4 5 Bonifacino, Juan S.; Glick, Benjamin S. (2004-01-23). "The Mechanisms of Vesicle Budding and Fusion". Cell. 116 (2): 153–166. doi: 10.1016/S0092-8674(03)01079-1 . ISSN 0092-8674. PMID 14744428.
- ↑ Hsu, Victor W.; Yang, Jia-Shu (2009-12-03). "Mechanisms of COPI vesicle formation". FEBS Letters. 583 (23): 3758–3763. doi:10.1016/j.febslet.2009.10.056. ISSN 0014-5793. PMC 2788077 . PMID 19854177.
- 1 2 3 Sato, Ken; Nakano, Akihiko (2007-05-22). "Mechanisms of COPII vesicle formation and protein sorting". FEBS Letters. Membrane Trafficking. 581 (11): 2076–2082. doi: 10.1016/j.febslet.2007.01.091 . ISSN 0014-5793. PMID 17316621.
- 1 2 3 4 Lajtha, Abel. (2010). Handbook of Neurochemistry and Molecular Neurobiology. Springer Verlag. ISBN 978-0-387-35443-9. OCLC 462919553.
- 1 2 3 4 5 Russo, Roberta; Esposito, Maria Rosaria; Iolascon, Achille (2013). "Inherited hematological disorders due to defects in coat protein (COP)II complex". American Journal of Hematology. 88 (2): 135–140. doi: 10.1002/ajh.23292 . ISSN 1096-8652. PMID 22764119.
- 1 2 3 4 Heimpel, Hermann; Anselstetter, Volker; Chrobak, Ladislav; Denecke, Jonas; Einsiedler, Beate; Gallmeier, Kerstin; Griesshammer, Antje; Marquardt, Thorsten; Janka-Schaub, Gritta; Kron, Martina; Kohne, Elisabeth (2003-12-15). "Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation". Blood. 102 (13): 4576–4581. doi:10.1182/blood-2003-02-0613. ISSN 0006-4971. PMID 12933587. S2CID 1553686.
- 1 2 3 4 Spreafico, Marta; Peyvandi, Flora (June 2009). "Combined Factor V and Factor VIII Deficiency". Seminars in Thrombosis and Hemostasis. 35 (4): 390–399. doi:10.1055/s-0029-1225761. ISSN 1098-9064. PMID 19598067.