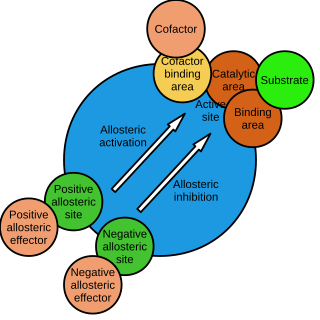
In biochemistry, allosteric regulation is the regulation of an enzyme by binding an effector molecule at a site other than the enzyme's active site.
Within the fields of molecular biology and pharmacology, a small molecule or micromolecule is a low molecular weight organic compound that may regulate a biological process, with a size on the order of 1 nm. Many drugs are small molecules; the terms are equivalent in the literature. Larger structures such as nucleic acids and proteins, and many polysaccharides are not small molecules, although their constituent monomers are often considered small molecules. Small molecules may be used as research tools to probe biological function as well as leads in the development of new therapeutic agents. Some can inhibit a specific function of a protein or disrupt protein–protein interactions.
Chemical specificity is the ability of binding site of a macromolecule to bind specific ligands. The fewer ligands a protein can bind, the greater its specificity.
In biochemistry and molecular biology, a binding site is a region on a macromolecule such as a protein that binds to another molecule with specificity. The binding partner of the macromolecule is often referred to as a ligand. Ligands may include other proteins, enzyme substrates, second messengers, hormones, or allosteric modulators. The binding event is often, but not always, accompanied by a conformational change that alters the protein's function. Binding to protein binding sites is most often reversible, but can also be covalent reversible or irreversible.

In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
A biological target is anything within a living organism to which some other entity is directed and/or binds, resulting in a change in its behavior or function. Examples of common classes of biological targets are proteins and nucleic acids. The definition is context-dependent, and can refer to the biological target of a pharmacologically active drug compound, the receptor target of a hormone, or some other target of an external stimulus. Biological targets are most commonly proteins such as enzymes, ion channels, and receptors.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
In biology, cell signaling or cell communication is the ability of a cell to receive, process, and transmit signals with its environment and with itself. Cell signaling is a fundamental property of all cellular life in prokaryotes and eukaryotes. Signals that originate from outside a cell can be physical agents like mechanical pressure, voltage, temperature, light, or chemical signals. Chemical signals can be hydrophobic or hydrophilic. Cell signaling can occur over short or long distances, and as a result can be classified as autocrine, juxtacrine, intracrine, paracrine, or endocrine. Signaling molecules can be synthesized from various biosynthetic pathways and released through passive or active transports, or even from cell damage.

In pharmacology, the term mechanism of action (MOA) refers to the specific biochemical interaction through which a drug substance produces its pharmacological effect. A mechanism of action usually includes mention of the specific molecular targets to which the drug binds, such as an enzyme or receptor. Receptor sites have specific affinities for drugs based on the chemical structure of the drug, as well as the specific action that occurs there.

An enzyme inhibitor is a molecule that binds to an enzyme and blocks its activity. Enzymes are proteins that speed up chemical reactions necessary for life, in which substrate molecules are converted into products. An enzyme facilitates a specific chemical reaction by binding the substrate to its active site, a specialized area on the enzyme that accelerates the most difficult step of the reaction.
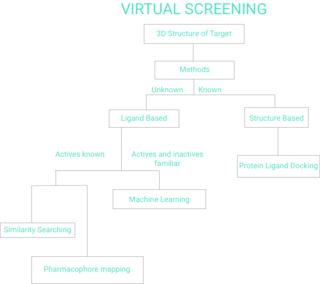
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
High-content screening (HCS), also known as high-content analysis (HCA) or cellomics, is a method that is used in biological research and drug discovery to identify substances such as small molecules, peptides, or RNAi that alter the phenotype of a cell in a desired manner. Hence high content screening is a type of phenotypic screen conducted in cells involving the analysis of whole cells or components of cells with simultaneous readout of several parameters. HCS is related to high-throughput screening (HTS), in which thousands of compounds are tested in parallel for their activity in one or more biological assays, but involves assays of more complex cellular phenotypes as outputs. Phenotypic changes may include increases or decreases in the production of cellular products such as proteins and/or changes in the morphology of the cell. Hence HCA typically involves automated microscopy and image analysis. Unlike high-content analysis, high-content screening implies a level of throughput which is why the term "screening" differentiates HCS from HCA, which may be high in content but low in throughput.
Fragment-based lead discovery (FBLD) also known as fragment-based drug discovery (FBDD) is a method used for finding lead compounds as part of the drug discovery process. Fragments are small organic molecules which are small in size and low in molecular weight. It is based on identifying small chemical fragments, which may bind only weakly to the biological target, and then growing them or combining them to produce a lead with a higher affinity. FBLD can be compared with high-throughput screening (HTS). In HTS, libraries with up to millions of compounds, with molecular weights of around 500 Da, are screened, and nanomolar binding affinities are sought. In contrast, in the early phase of FBLD, libraries with a few thousand compounds with molecular weights of around 200 Da may be screened, and millimolar affinities can be considered useful. FBLD is a technique being used in research for discovering novel potent inhibitors. This methodology could help to design multitarget drugs for multiple diseases. The multitarget inhibitor approach is based on designing an inhibitor for the multiple targets. This type of drug design opens up new polypharmacological avenues for discovering innovative and effective therapies. Neurodegenerative diseases like Alzheimer’s (AD) and Parkinson’s, among others, also show rather complex etiopathologies. Multitarget inhibitors are more appropriate for addressing the complexity of AD and may provide new drugs for controlling the multifactorial nature of AD, stopping its progression.

Cell surface receptors are receptors that are embedded in the plasma membrane of cells. They act in cell signaling by receiving extracellular molecules. They are specialized integral membrane proteins that allow communication between the cell and the extracellular space. The extracellular molecules may be hormones, neurotransmitters, cytokines, growth factors, cell adhesion molecules, or nutrients; they react with the receptor to induce changes in the metabolism and activity of a cell. In the process of signal transduction, ligand binding affects a cascading chemical change through the cell membrane.
Druggability is a term used in drug discovery to describe a biological target that is known to or is predicted to bind with high affinity to a drug. Furthermore, by definition, the binding of the drug to a druggable target must alter the function of the target with a therapeutic benefit to the patient. The concept of druggability is most often restricted to small molecules but also has been extended to include biologic medical products such as therapeutic monoclonal antibodies.
Chemical genetics is the investigation of the function of proteins and signal transduction pathways in cells by the screening of chemical libraries of small molecules. Chemical genetics is analogous to classical genetic screen where random mutations are introduced in organisms, the phenotype of these mutants is observed, and finally the specific gene mutation (genotype) that produced that phenotype is identified. In chemical genetics, the phenotype is disturbed not by introduction of mutations, but by exposure to small molecule tool compounds. Phenotypic screening of chemical libraries is used to identify drug targets or to validate those targets in experimental models of disease. Recent applications of this topic have been implicated in signal transduction, which may play a role in discovering new cancer treatments. Chemical genetics can serve as a unifying study between chemistry and biology. The approach was first proposed by Tim Mitchison in 1994 in an opinion piece in the journal Chemistry & Biology entitled "Towards a pharmacological genetics".
A thermal shift assay (TSA) measures changes in the thermal denaturation temperature and hence stability of a protein under varying conditions such as variations in drug concentration, buffer pH or ionic strength, redox potential, or sequence mutation. The most common method for measuring protein thermal shifts is differential scanning fluorimetry (DSF) or thermofluor, which utilizes specialized fluorogenic dyes.
Chemoproteomics entails a broad array of techniques used to identify and interrogate protein-small molecule interactions. Chemoproteomics complements phenotypic drug discovery, a paradigm that aims to discover lead compounds on the basis of alleviating a disease phenotype, as opposed to target-based drug discovery, in which lead compounds are designed to interact with predetermined disease-driving biological targets. As phenotypic drug discovery assays do not provide confirmation of a compound's mechanism of action, chemoproteomics provides valuable follow-up strategies to narrow down potential targets and eventually validate a molecule's mechanism of action. Chemoproteomics also attempts to address the inherent challenge of drug promiscuity in small molecule drug discovery by analyzing protein-small molecule interactions on a proteome-wide scale. A major goal of chemoproteomics is to characterize the interactome of drug candidates to gain insight into mechanisms of off-target toxicity and polypharmacology.
Target 2035 is a global effort or movement to discover open science, pharmacological modulator(s) for every protein in the human proteome by the year 2035. The effort is led by the Structural Genomics Consortium with the intention that this movement evolves organically. Target 2035 has been borne out of the success that chemical probes have had in elevating or de-prioritizing the therapeutic potential of protein targets. The availability of open access pharmacological tools is a largely unmet aspect of drug discovery especially for the dark proteome.
