
Haploinsufficiency in genetics describes a model of dominant gene action in diploid organisms, in which a single copy of the wild-type allele at a locus in heterozygous combination with a variant allele is insufficient to produce the wild-type phenotype. Haploinsufficiency may arise from a de novo or inherited loss-of-function mutation in the variant allele, such that it yields little or no gene product. Although the other, standard allele still produces the standard amount of product, the total product is insufficient to produce the standard phenotype. This heterozygous genotype may result in a non- or sub-standard, deleterious, and (or) disease phenotype. Haploinsufficiency is the standard explanation for dominant deleterious alleles.

In the inner ear, stereocilia are the mechanosensing organelles of hair cells, which respond to fluid motion in numerous types of animals for various functions, including hearing and balance. They are about 10–50 micrometers in length and share some similar features of microvilli. The hair cells turn the fluid pressure and other mechanical stimuli into electric stimuli via the many microvilli that make up stereocilia rods. Stereocilia exist in the auditory and vestibular systems.
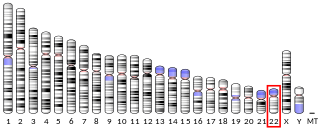
T-box transcription factor TBX1 also known as T-box protein 1 and testis-specific T-box protein is a protein that in humans is encoded by the TBX1 gene. Genes in the T-box family are transcription factors that play important roles in the formation of tissues and organs during embryonic development. To carry out these roles, proteins made by this gene family bind to specific areas of DNA called T-box binding element (TBE) to control the expression of target genes.
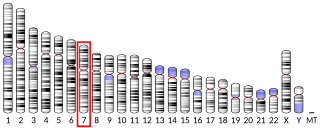
Pendrin is an anion exchange protein that in humans is encoded by the SLC26A4 gene . Pendrin was initially identified as a sodium-independent chloride-iodide exchanger with subsequent studies showing that it also accepts formate and bicarbonate as substrates. Pendrin is similar to the Band 3 transport protein found in red blood cells. Pendrin is the protein which is mutated in Pendred syndrome, which is an autosomal recessive disorder characterized by sensorineural hearing loss, goiter and a partial organification problem detectable by a positive perchlorate test.
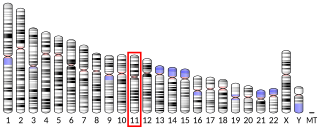
Harmonin is a protein that in humans is encoded by the USH1C gene. It is expressed in sensory cells of the inner ear and retina, where it plays a role in hearing, balance, and vision. Mutations at the USH1C locus cause Usher syndrome type 1c and nonsyndromic sensorineural deafness.

Cadherin-23 is a protein that in humans is encoded by the CDH23 gene.

Protocadherin-15 is a protein that in humans is encoded by the PCDH15 gene.

Gap junction beta-6 protein (GJB6), also known as connexin 30 (Cx30) — is a protein that in humans is encoded by the GJB6 gene. Connexin 30 (Cx30) is one of several gap junction proteins expressed in the inner ear. Mutations in gap junction genes have been found to lead to both syndromic and nonsyndromic deafness. Mutations in this gene are associated with Clouston syndrome.

Tyrosinase-related protein 1, also known as TYRP1, is an intermembrane enzyme which in humans is encoded by the TYRP1 gene.

P protein, also known as melanocyte-specific transporter protein or pink-eyed dilution protein homolog, is a protein that in humans is encoded by the oculocutaneous albinism II (OCA2) gene. The P protein is believed to be an integral membrane protein involved in small molecule transport, specifically of tyrosine—a precursor of melanin. Certain mutations in OCA2 result in type 2 oculocutaneous albinism. OCA2 encodes the human homologue of the mouse p gene.

Eyes absent homolog 1 is a protein that in humans is encoded by the EYA1 gene.
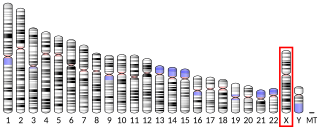
Mitochondrial import inner membrane translocase subunit Tim8 A, also known as deafness-dystonia peptide or protein is an enzyme that in humans is encoded by the TIMM8A gene. This translocase has similarity to yeast mitochondrial proteins that are involved in the import of metabolite transporters from the cytoplasm into the mitochondrial inner membrane. The gene is mutated in deafness-dystonia syndrome and it is postulated that MTS/DFN-1 is a mitochondrial disease caused by a defective mitochondrial protein import system.
Homeobox protein Hox-A13 is a protein that in humans is encoded by the HOXA13 gene.
Alpha-tectorin is a protein that in humans is encoded by the TECTA gene.

Eyes absent homolog 4 is a protein that in humans is encoded by the EYA4 gene.

Otoferlin is a protein that in humans is encoded by the OTOF gene. It is involved in vesicle membrane fusion, and mutations in the OTOF gene are associated with a genetic form of deafness.
Unconventional myosin-XV is a protein that in humans is encoded by the MYO15A gene.
Special AT-rich sequence-binding protein 2 (SATB2) also known as DNA-binding protein SATB2 is a protein that in humans is encoded by the SATB2 gene. SATB2 is a DNA-binding protein that specifically binds nuclear matrix attachment regions and is involved in transcriptional regulation and chromatin remodeling. SATB2 shows a restricted mode of expression and is expressed in certain cell nuclei. The SATB2 protein is mainly expressed in the epithelial cells of the colon and rectum, followed by the nuclei of neurons in the brain.

Espin, also known as autosomal recessive deafness type 36 protein or ectoplasmic specialization protein, is a protein that in humans is encoded by the ESPN gene. Espin is a microfilament binding protein.
Otoancorin is a protein found in the vertebrate inner ear, on the sensory epithelia where it connects to the gel matrix.



