Related Research Articles
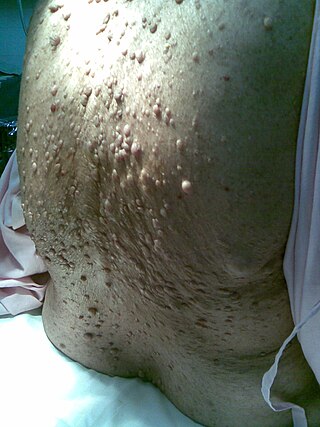
Neurofibromatosis (NF) refers to a group of three distinct genetic conditions in which tumors grow in the nervous system. The tumors are non-cancerous (benign) and often involve the skin or surrounding bone. Although symptoms are often mild, each condition presents differently. Neurofibromatosis type I (NF1) is typically characterized by café au lait spots, neurofibromas, scoliosis, and headaches. Neurofibromatosis type II (NF2), on the other hand, may present with early-onset hearing loss, cataracts, tinnitus, difficulty walking or maintain balance, and muscle atrophy. The third type is called schwannomatosis and often presents in early adulthood with widespread pain, numbness, or tingling due to nerve compression.
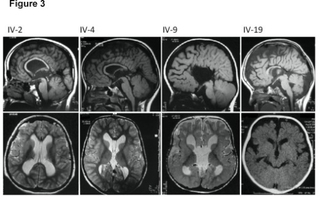
Megalencephaly is a growth development disorder in which the brain is abnormally large. It is characterized by a brain with an average weight that is 2.5 standard deviations above the mean of the general population. Approximately 1 out of 50 children (2%) are said to have the characteristics of megalencephaly in the general population.

Cardiofaciocutaneous (CFC) syndrome is an extremely rare genetic disorder, and is one of the RASopathies. It was first described in 1986.
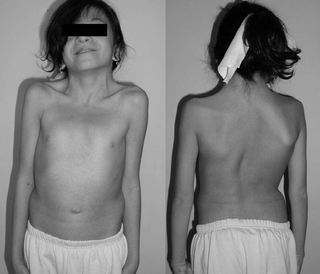
Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations. Facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw. Heart problems may include pulmonary valve stenosis. The breast bone may either protrude or be sunken, while the spine may be abnormally curved. Intelligence is often normal. Complications of NS can include leukemia.
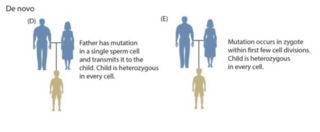
A germline mutation, or germinal mutation, is any detectable variation within germ cells. Mutations in these cells are the only mutations that can be passed on to offspring, when either a mutated sperm or oocyte come together to form a zygote. After this fertilization event occurs, germ cells divide rapidly to produce all of the cells in the body, causing this mutation to be present in every somatic and germline cell in the offspring; this is also known as a constitutional mutation. Germline mutation is distinct from somatic mutation.

Neurofibromatosis type I (NF-1), or von Recklinghausen syndrome, is a complex multi-system human disorder caused by the mutation of neurofibromin 1 (NF-1), a gene on chromosome 17 that is responsible for production of a protein (neurofibromin) which is needed for normal function in many human cell types. NF-1 causes tumors along the nervous system which can grow anywhere on the body. NF-1 is one of the most common genetic disorders and is not limited to any person's race or sex. NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1.

Cowden syndrome is an autosomal dominant inherited condition characterized by benign overgrowths called hamartomas as well as an increased lifetime risk of breast, thyroid, uterine, and other cancers. It is often underdiagnosed due to variability in disease presentation, but 99% of patients report mucocutaneous symptoms by age 20–29. Despite some considering it a primarily dermatologic condition, Cowden's syndrome is a multi-system disorder that also includes neurodevelopmental disorders such as macrocephaly.
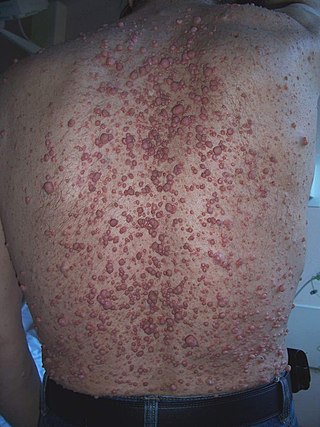
A neurofibroma is a benign nerve-sheath tumor in the peripheral nervous system. In 90% of cases, they are found as stand-alone tumors, while the remainder are found in persons with neurofibromatosis type I (NF1), an autosomal-dominant genetically inherited disease. They can result in a range of symptoms from physical disfiguration and pain to cognitive disability.
Costello syndrome, also called faciocutaneoskeletal syndrome or FCS syndrome, is a rare genetic disorder that affects many parts of the body. It is characterized by delayed development and intellectual disabilities, distinctive facial features, unusually flexible joints, and loose folds of extra skin, especially on the hands and feet. Heart abnormalities are common, including a very fast heartbeat (tachycardia), structural heart defects, and overgrowth of the heart muscle. Infants with Costello syndrome may be large at birth, but grow more slowly than other children and have difficulty feeding. Later in life, people with this condition have relatively short stature and many have reduced levels of growth hormones. It is a RASopathy.

GTPase HRas, from "Harvey Rat sarcoma virus", also known as transforming protein p21 is an enzyme that in humans is encoded by the HRAS gene. The HRAS gene is located on the short (p) arm of chromosome 11 at position 15.5, from base pair 522,241 to base pair 525,549. HRas is a small G protein in the Ras subfamily of the Ras superfamily of small GTPases. Once bound to Guanosine triphosphate, H-Ras will activate a Raf kinase like c-Raf, the next step in the MAPK/ERK pathway.

Noonan syndrome with multiple lentigines (NSML) which is part of a group called Ras/MAPK pathway syndromes, is a rare autosomal dominant, multisystem disease caused by a mutation in the protein tyrosine phosphatase, non-receptor type 11 gene (PTPN11). The disease is a complex of features, mostly involving the skin, skeletal and cardiovascular systems, which may or may not be present in all patients. The nature of how the mutation causes each of the condition's symptoms is not well known; however, research is ongoing. It is a RASopathy.

Neurofibromin (NF-1) is a protein that is encoded in the human by the NF1 gene. NF1 is located on chromosome 17. Neurofibromin, a GTPase-activating protein that negatively regulates RAS/MAPK pathway activity by accelerating the hydrolysis of Ras-bound GTP. NF1 has a high mutation rate and mutations can alter cellular growth control, and neural development, resulting in neurofibromatosis type 1. Symptoms of NF1 include disfiguring cutaneous neurofibromas (CNF), café au lait pigment spots, plexiform neurofibromas (PN), skeletal defects, optic nerve gliomas, life-threatening malignant peripheral nerve sheath tumors (MPNST), pheochromocytoma, attention deficits, learning deficits and other cognitive disabilities.

Synaptic Ras GTPase-activating protein 1, also known as synaptic Ras-GAP 1 or SYNGAP1, is a protein that in humans is encoded by the SYNGAP1 gene. SYNGAP1 is a ras GTPase-activating protein that is critical for the development of cognition and proper synapse function. Mutations in humans can cause intellectual disability, epilepsy, autism and sensory processing deficits.
Leucine-zipper-like transcriptional regulator 1 is a protein that in humans is encoded by the LZTR1 gene.
Germline mosaicism, also called gonadal mosaicism, is a type of genetic mosaicism where more than one set of genetic information is found specifically within the gamete cells; conversely, somatic mosaicism is a type of genetic mosaicism found in somatic cells. Germline mosaicism can be present at the same time as somatic mosaicism or individually, depending on when the conditions occur. Pure germline mosaicism refers to mosaicism found exclusively in the gametes and not in any somatic cells. Germline mosaicism can be caused either by a mutation that occurs after conception, or by epigenetic regulation, alterations to DNA such as methylation that do not involve changes in the DNA coding sequence.
Parkes Weber syndrome (PWS) is a congenital disorder of the vascular system. It is an extremely rare condition, and its exact prevalence is unknown. It is named after British dermatologist Frederick Parkes Weber, who first described the syndrome in 1907.

Macrocephaly-capillary malformation (M-CM) is a multiple malformation syndrome causing abnormal body and head overgrowth and cutaneous, vascular, neurologic, and limb abnormalities. Though not every patient has all features, commonly found signs include macrocephaly, congenital macrosomia, extensive cutaneous capillary malformation, body asymmetry, polydactyly or syndactyly of the hands and feet, lax joints, doughy skin, variable developmental delay and other neurologic problems such as seizures and low muscle tone.
Legius syndrome (LS) is an autosomal dominant condition characterized by cafe au lait spots. It was first described in 2007 and is often mistaken for neurofibromatosis type I. It is caused by mutations in the SPRED1 gene. It is also known as neurofibromatosis type 1-like syndrome.
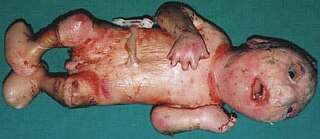
Neu–Laxova syndrome is a rare autosomal recessive disorder characterized by severe intrauterine growth restriction and multiple congenital malformations. Neu–Laxova syndrome is a very severe disorder, leading to stillbirth or death shortly after birth. It was first described by Dr. Richard Neu in 1971 and Dr. Renata Laxova in 1972 as a lethal disorder in siblings with multiple malformations. Neu–Laxova syndrome is an extremely rare disorder with fewer than 100 cases reported in medical literature.
SYNGAP1-related intellectual disability is a monogenetic developmental and epileptic encephalopathy that affects the central nervous system. Symptoms include intellectual disability, epilepsy, autism, sensory processing deficits, hypotonia and unstable gait.
References
- 1 2 3 Rauen KA (2022). "Defining RASopathy". Disease Models & Mechanisms. 15 (2). doi:10.1242/dmm.049344. PMC 8821523 . PMID 35103797.
- 1 2 Tidyman WE, Rauen KA (2016). "Pathogenetics of the RASopathies". Human Molecular Genetics. 25 (R2): R123–R132. doi:10.1093/hmg/ddw191. PMC 6283265 . PMID 27412009.
- ↑ Riller Q, Rieux-Laucat F (2021). "RASopathies: From germline mutations to somatic and multigenic diseases". Biomedical Journal. 44 (4): 422–432. doi:10.1016/j.bj.2021.06.004. PMC 8514848 . PMID 34175492.
- ↑ Dunnett-Kane V, Burkitt-Wright E, Blackhall FH, Malliri A, Evans DG, Lindsay CR (2020). "Germline and sporadic cancers driven by the RAS pathway: parallels and contrasts". Annals of Oncology. 31 (7): 873–883. doi:10.1016/j.annonc.2020.03.291. PMC 7322396 . PMID 32240795.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Rai B, Naylor PE, Siqueiros-Sanchez M, Wintermark M, Raman MM, Jo B; et al. (2023). "Novel effects of Ras-MAPK pathogenic variants on the developing human brain and their link to gene expression and inhibition abilities". Translational Psychiatry. 13 (1): 245. doi:10.1038/s41398-023-02504-4. PMC 10322993 . PMID 37407569.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Zenker M (2022). "Clinical overview on RASopathies". American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 190 (4): 414–424. doi: 10.1002/ajmg.c.32015 . PMID 36428239.
- ↑ Aoki Y, Niihori T, Inoue S, Matsubara Y (2016). "Recent advances in RASopathies". Journal of Human Genetics. 61 (1): 33–9. doi:10.1038/jhg.2015.114. PMID 26446362.
{{cite journal}}: CS1 maint: multiple names: authors list (link)