
Dystrophin is a rod-shaped cytoplasmic protein, and a vital part of a protein complex that connects the cytoskeleton of a muscle fiber to the surrounding extracellular matrix through the cell membrane. This complex is variously known as the costamere or the dystrophin-associated protein complex (DAPC). Many muscle proteins, such as α-dystrobrevin, syncoilin, synemin, sarcoglycan, dystroglycan, and sarcospan, colocalize with dystrophin at the costamere. It has a molecular weight of 427 kDa
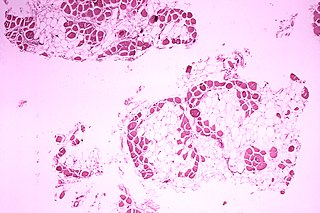
Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy predominantly affecting boys. The onset of muscle weakness typically begins around age four, with rapid progression. Initially, muscle loss occurs in the thighs and pelvis, extending to the arms, which can lead to difficulties in standing up. By the age of 12, most individuals with Duchenne muscular dystrophy are unable to walk. Affected muscles may appear larger due to an increase in fat content, and scoliosis is common. Some individuals may experience intellectual disability, and females carrying a single copy of the mutated gene may show mild symptoms.
Antisense therapy is a form of treatment that uses antisense oligonucleotides (ASOs) to target messenger RNA (mRNA). ASOs are capable of altering mRNA expression through a variety of mechanisms, including ribonuclease H mediated decay of the pre-mRNA, direct steric blockage, and exon content modulation through splicing site binding on pre-mRNA. Several ASOs have been approved in the United States, the European Union, and elsewhere.
Sarepta Therapeutics, Inc. is a medical research and drug development company with corporate offices and research facilities in Cambridge, Massachusetts, United States. Incorporated in 1980 as AntiVirals, shortly before going public the company changed its name from AntiVirals to AVI BioPharma soon with stock symbol AVII and in July 2012 changed name from AVI BioPharma to Sarepta Therapeutics and SRPT respectively. As of 2023, the company has four approved drugs.
The dystrophin-associated protein complex, also known as the dystrophin-associated glycoprotein complex is a multiprotein complex that includes dystrophin and the dystrophin-associated proteins. It is one of the two protein complexes that make up the costamere in striated muscle cells. The other complex is the integrin-vinculin-talin complex.

Deflazacort is a glucocorticoid belonging to acetonides or O-isopropylidene derivative. It is used as an anti-inflammatory and was patented in 1969 and approved for medical use in 1985. The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication for Duchenne Muscular Dystrophy.
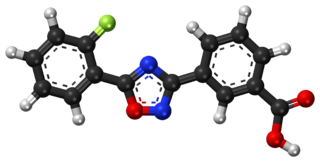
Ataluren, sold under the brand name Translarna, is a medication for the treatment of Duchenne muscular dystrophy. It was designed by PTC Therapeutics.
In molecular biology, exon skipping is a form of RNA splicing used to cause cells to “skip” over faulty or misaligned sections (exons) of genetic code, leading to a truncated but still functional protein despite the genetic mutation.
Prosensa was a biotechnology company engaged in the discovery, development and commercialization of RNA-modulating therapeutics. The company targets genetic disorders with a large unmet medical need, with a primary focus on neuromuscular and neurodegenerative disorders such as Duchenne muscular dystrophy (DMD), myotonic dystrophy, and Huntington's disease. Prosensa was acquired by BioMarin
Drisapersen is an experimental drug that was under development by BioMarin, after acquisition of Prosensa, for the treatment of Duchenne muscular dystrophy. The drug is a 2'-O-methyl phosphorothioate oligonucleotide that alters the splicing of the dystrophin RNA transcript, eliminating exon 51 from the mature dystrophin mRNA.
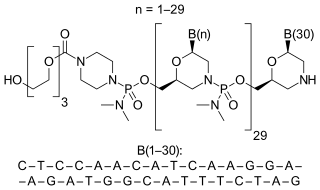
Eteplirsen is a medication to treat, but not cure, some types of Duchenne muscular dystrophy (DMD), caused by a specific mutation. Eteplirsen only targets specific mutations and can be used to treat about 14% of DMD cases. Eteplirsen is a form of antisense therapy.

Marathon Pharmaceuticals LLC was a privately held biopharmaceuticals company focused on drugs for people with rare diseases. The Illinois-based company developed and manufactured therapeutics and brought them to market. It employed 100 people in four global locations. In 2017, PTC Therapeutics acquired rights to Marathon Pharmaceuticals' drug Emflaza (deflazacort) for $140 million after criticism about their plan to sell the drug at a list price of $89,000 per year to sufferers despite the fact that the same drug was available in Canada and the UK for around $1,000 per year.

Ezutromid is an orally administered small molecule utrophin modulator involved in a Phase 2 clinical trial produced by Summit Therapeutics for the treatment of Duchenne muscular dystrophy (DMD). DMD is a fatal x-linked recessive disease affecting approximately 1 in 5000 males and is a designated orphan disease by the FDA and European Medicines Agency. Approximately 1/3 of the children obtain DMD as a result of spontaneous mutation in the dystrophin gene and have no family history of the disease. Dystrophin is a vital component of mature muscle function, and therefore DMD patients have multifarious forms of defunct or deficient dystrophin proteins that all manifest symptomatically as muscle necrosis and eventually organ failure. Ezutromid is theorized to maintain utrophin, a protein functionally and structurally similar to dystrophin that precedes and is replaced by dystrophin during development. Utrophin and dystrophin are reciprocally expressed, and are found in different locations in a mature muscle cell. However, in dystrophin-deficient patients, utrophin was found to be upregulated and is theorized to replace dystrophin in order to maintain muscle fibers. Ezutromid is projected to have the potential to treat all patients suffering with DMD as it maintains the production of utrophin to counteract the lack of dystrophin to retard muscle degeneration. Both the FDA and European Medicines Agency has given ezutromid an orphan drug designation. The FDA Office of Orphan Products and Development offers an Orphan Drug Designation program (ODD) that allows drugs aimed to treat diseases that affect less than 200,000 people in the U.S. monetary incentives such as a period of market exclusivity, tax incentives, and expedited approval processes.

Vamorolone, sold under the brand name Agamree, is a synthetic corticosteroid, which is used for the treatment of Duchenne muscular dystrophy. It is taken by mouth. It is a dual atypical glucocorticoid and antimineralocorticoid.

Viltolarsen, sold under the brand name Viltepso, is a medication used for the treatment of Duchenne muscular dystrophy (DMD). Viltolarsen is a Morpholino antisense oligonucleotide.

Cure Rare Disease is a non-profit biotechnology company based in Boston, Massachusetts that is working to create novel therapeutics using gene therapy, gene editing and antisense oligonucleotides to treat people impacted by rare and ultra-rare genetic neuromuscular conditions.
Toshifumi (Toshi) Yokota is a biomedical scientist and professor of medical genetics at the University of Alberta, also holding the titles of the Friends of Garrett Cumming Research & Muscular Dystrophy Canada Endowed Research Chair and the Henri M. Toupin Chair in Neurological Science. Known for pioneering research in antisense therapy for muscular dystrophy that led to the development of an FDA-approved drug viltolarsen, research interests encompass precision medicine for muscular dystrophy and genetic diseases. Publications exceed 100 refereed papers and patents, with contributions as co-editor to three books in the Methods in Molecular Biology series from Humana Press, Springer-Nature, Roles include fellow of the Canadian Academy of Health Sciences, a member of the editorial boards for numerous journals, a member of the Medical and Scientific Advisory Committee of Muscular Dystrophy Canada, chief scientific officer of OligomicsTx, and a co-founder of the Canadian Neuromuscular Network (CAN-NMD).
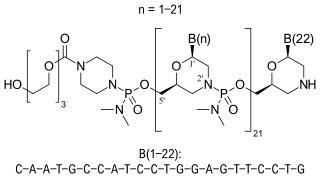
Casimersen, sold under the brand name Amondys 45, is an antisense oligonucleotide medication used for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the dystrophin gene that is amenable to exon 45 skipping. It is an antisense oligonucleotide of phosphorodiamidate morpholino oligomer (PMO). Duchenne muscular dystrophy is a rare disease that primarily affects boys. It is caused by low levels of a muscle protein called dystrophin. The lack of dystrophin causes progressive muscle weakness and premature death.
Delandistrogene moxeparvovec, sold under the brand name Elevidys, is a recombinant gene therapy used for the treatment of Duchenne muscular dystrophy. It is designed to deliver into the body a gene that leads to production of Elevidys micro-dystrophin that contains selected domains of the dystrophin protein present in normal muscle cells. It is an adeno-associated virus vector-based gene therapy that is given by injection into a vein.
Stephen Donald Wilton, also known as Steve Wilton, is an Australian molecular biologist and academic, serving as the Foundation Professor of Molecular Therapy at Murdoch University and adjunct professor at the University of Western Australia (UWA). He also fulfills dual roles as a Director at the Perron Institute for Neurological and Translational Science and deputy director at Murdoch's Centre for Molecular Medicine and Innovative Therapeutics (CMMIT).
