
A dipeptide is an organic compound derived from two amino acids. The constituent amino acids can be the same or different. When different, two isomers of the dipeptide are possible, depending on the sequence. Several dipeptides are physiologically important, and some are both physiologically and commercially significant. A well known dipeptide is aspartame, an artificial sweetener.
Alpha-1 blockers constitute a variety of drugs that block the effect of catecholamines on alpha-1-adrenergic receptors. They are mainly used to treat benign prostatic hyperplasia (BPH), hypertension and post-traumatic stress disorder. Alpha-1-adrenergic receptors are present in vascular smooth muscle, the central nervous system, and other tissues. When alpha blockers bind to these receptors in vascular smooth muscle, they cause vasodilation.
Neurokinin 1 (NK1) antagonists (-pitants) are a novel class of medications that possesses unique antidepressant, anxiolytic, and antiemetic properties. NK-1 antagonists boost the efficacy of 5-HT3 antagonists to prevent nausea and vomiting. The discovery of neurokinin 1 (NK1) receptor antagonists was a turning point in the prevention of nausea and vomiting associated with cancer chemotherapy.

The oxytocin receptor, also known as OXTR, is a protein which functions as receptor for the hormone and neurotransmitter oxytocin. In humans, the oxytocin receptor is encoded by the OXTR gene which has been localized to human chromosome 3p25.

Silodosin, sold under the brand name Urief among others, is a medication for the symptomatic treatment of benign prostatic hyperplasia. It acts as an alpha-1 adrenergic receptor antagonist.

Propiram is a partial μ-opioid receptor agonist and weak μ antagonist analgesic from the ampromide family of drugs related to other drugs such as phenampromide and diampromide. It was invented in 1963 in the United Kingdom by Bayer but was not widely marketed, although it saw some limited clinical use, especially in dentistry. Propiram reached Phase III clinical trials in the United States and Canada.

Renin inhibitors are pharmaceutical drugs inhibiting the activity of renin that is responsible for hydrolyzing angiotensinogen to angiotensin I, which in turn reduces the formation of angiotensin II that facilitates blood pressure.

L-371,257 is a compound used in scientific research which acts as a selective antagonist of the oxytocin receptor with over 800x selectivity over the related vasopressin receptors. It was one of the first non-peptide oxytocin antagonists developed, and has good oral bioavailability, but poor penetration of the blood–brain barrier, which gives it good peripheral selectivity with few central side effects. Potential applications are likely to be in the treatment of premature labour.

WAY-267464 is a potent, selective, non-peptide agonist for the oxytocin receptor, with negligible affinity for the vasopressin receptors. Contradictorily however, though originally described as selective for the oxytocin receptor and lacking affinity for the vasopressin receptors, it has since been reported to also act as a potent vasopressin V1A receptor antagonist. WAY-267464 has been shown to cross the blood–brain barrier to a significantly greater extent than exogenously applied oxytocin, and in animal tests produces centrally-mediated oxytocinergic actions such as anxiolytic effects, but with no antidepressant effect evident. It was developed by a team at Ferring Pharmaceuticals. WAY-267464 was under investigation for the potential clinical treatment of anxiety disorders by Wyeth, and reached the preclinical stage of development, but no development has been reported as of 2011.

L-368,899 is a drug used in scientific research which acts as a selective antagonist of the oxytocin receptor, with good selectivity over the related vasopressin receptors. Unlike related drugs such as the peripherally selective L-371,257, the oral bioavailabity is high and the brain penetration of L-368,899 is rapid, with selective accumulation in areas of the limbic system. This makes it a useful tool for investigating the centrally mediated roles of oxytocin, such as in social behaviour and pair bonding, and studies in primates have shown L-368,899 to reduce a number of behaviours such as food sharing, sexual activity and caring for infants, demonstrating the importance of oxytocinergic signalling in mediating these important social behaviours.

Epristeride, sold under the brand names Aipuliete and Chuanliu, is a medication which is used in the treatment of enlarged prostate in China. It is taken by mouth.

Retosiban also known as GSK-221,149-A is an oral drug which acts as an oxytocin receptor antagonist. It is being developed by GlaxoSmithKline for the treatment of preterm labour. Retosiban has high affinity for the oxytocin receptor and has greater than 1400-fold selectivity over the related vasopressin receptors
2,5-Diketopiperazine is an organic compound with the formula (NHCH2C(O))2. The compound features a six-membered ring containing two amide groups at opposite positions in the ring. It was first compound containing a peptide bond to be characterized by X-ray crystallography in 1938. It is the parent of a large class of 2,5-Diketopiperazines (2,5-DKPs) with the formula (NHCH2(R)C(O))2 (R = H, CH3, etc.). They are ubiquitous peptide in nature. They are often found in fermentation broths and yeast cultures as well as embedded in larger more complex architectures in a variety of natural products as well as several drugs. In addition, they are often produced as degradation products of polypeptides, especially in processed foods and beverages. They have also been identified in the contents of comets.
A diketopiperazine (DKP), also known as a dioxopiperazine or piperazinedione, is a class of organic compounds related to piperazine but containing two amide linkages. DKP's are the smallest known class of cyclic peptide. Despite their name, they are not ketones, but amides. Three regioisomers are possible, differing in the locations of the carbonyl groups.
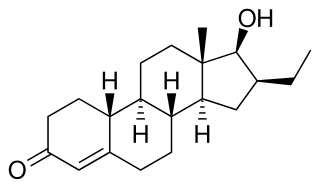
Oxendolone, sold under the brand names Prostetin and Roxenone, is an antiandrogen and progestin medication which is used in Japan in the treatment of enlarged prostate. However, this use is controversial due to concerns about its clinical efficacy. Oxendolone is not effective by mouth and must be given by injection into muscle.

Barusiban (INN) is a non-peptide drug which is among the most potent and selective oxytocin receptor antagonists known. It was trialed by Ferring Pharmaceuticals as a treatment of preterm labor but failed to demonstrate effectiveness and was not pursued any further.

A nonsteroidal antiandrogen (NSAA) is an antiandrogen with a nonsteroidal chemical structure. They are typically selective and full or silent antagonists of the androgen receptor (AR) and act by directly blocking the effects of androgens like testosterone and dihydrotestosterone (DHT). NSAAs are used in the treatment of androgen-dependent conditions in men and women. They are the converse of steroidal antiandrogens (SAAs), which are antiandrogens that are steroids and are structurally related to testosterone.

LG-120907 is a nonsteroidal antiandrogen (NSAA) of the quinoline group which was developed by Ligand Pharmaceuticals along with selective androgen receptor modulators (SARMs) like LG-121071 and was never marketed. The drug is a high-affinity antagonist of the androgen receptor (AR) with a Ki value of 26 nM and has been found to inhibit growth of the ventral prostate and seminal vesicles in male rats without increasing circulating levels of luteinizing hormone or testosterone. However, this tissue selectivity has not been assessed in humans. LG-120907 is orally active and shows greater oral potency than the arylpropionamide NSAA flutamide.

LIT-001 is a small-molecule oxytocin receptor agonist and vasopressin receptor mixed agonist and antagonist that was first described in the literature in 2018. Along with TC OT 39 and WAY-267464, it is one of the first small-molecule oxytocin receptor agonists to have been developed. LIT-001 has greatly improved pharmacokinetic properties relative to oxytocin, reduces social deficits in animal models, and may have potential as a therapeutic agent in the treatment of social disorders like autism in humans.


