Hypothesis that aging is caused by accumulated DNA damage
The DNA damage theory of aging proposes that aging is a consequence of unrepaired accumulation of naturally occurring DNA damage. Damage in this context is a DNA alteration that has an abnormal structure. Although both mitochondrial and nuclear DNA damage can contribute to aging, nuclear DNA is the main subject of this analysis. Nuclear DNA damage can contribute to aging either indirectly (by increasing apoptosis or cellular senescence) or directly (by increasing cell dysfunction).[1][2][3][4]
Several review articles have shown that deficient DNA repair, allowing greater accumulation of DNA damage, causes premature aging; and that increased DNA repair facilitates greater longevity, e.g.[5][6] Mouse models of nucleotide-excision–repair syndromes reveal a striking correlation between the degree to which specific DNA repair pathways are compromised and the severity of accelerated aging, strongly suggesting a causal relationship.[7] Human population studies show that single-nucleotide polymorphisms in DNA repair genes, causing up-regulation of their expression, correlate with increases in longevity.[8] Lombard et al. compiled a lengthy list of mouse mutational models with pathologic features of premature aging, all caused by different DNA repair defects.[9] Freitas and de Magalhães presented a comprehensive review and appraisal of the DNA damage theory of aging, including a detailed analysis of many forms of evidence linking DNA damage to aging.[2] As an example, they described a study showing that centenarians of 100 to 107 years of age had higher levels of two DNA repair enzymes, PARP1 and Ku70, than general-population old individuals of 69 to 75 years of age.[10][2] Their analysis supported the hypothesis that improved DNA repair leads to longer life span. Overall, they concluded that while the complexity of responses to DNA damage remains only partly understood, the idea that DNA damage accumulation with age is the primary cause of aging remains an intuitive and powerful one.[2]
In humans and other mammals, DNA damage occurs frequently and DNA repair processes have evolved to compensate.[11] In estimates made for mice, DNA lesions occur on average 25 to 115 times per minute in each cell, or about 36,000 to 160,000 per cell per day.[12] Some DNA damage may remain in any cell despite the action of repair processes. The accumulation of unrepaired DNA damage is more prevalent in certain types of cells, particularly in non-replicating or slowly replicating cells, such as cells in the brain, skeletal and cardiac muscle.[13]
To understand the DNA damage theory of aging it is important to distinguish between DNA damage and mutation, the two major types of errors that occur in DNA. Damage and mutation are fundamentally different. DNA damage is any physical abnormality in the DNA, such as single and double strand breaks, 8-hydroxydeoxyguanosine residues and polycyclic aromatic hydrocarbon adducts. DNA damage can be recognized by enzymes, and thus can be correctly repaired using the complementary undamaged strand in DNA as a template or an undamaged sequence in a homologous chromosome if it is available for copying. If a cell retains DNA damage, transcription of a gene can be prevented and thus translation into a protein will also be blocked. Replication may also be blocked and/or the cell may die. Descriptions of reduced function, characteristic of aging and associated with accumulation of DNA damage, are described in the next section.
In contrast to DNA damage, a mutation is a change in the base sequence of the DNA. A mutation cannot be recognized by enzymes once the base change is present in both DNA strands, and thus a mutation cannot be repaired. At the cellular level, mutations can cause alterations in protein function and regulation. Mutations are replicated when the cell replicates. In a population of cells, mutant cells will increase or decrease in frequency according to the effects of the mutation on the ability of the cell to survive and reproduce. Although distinctly different from each other, DNA damages and mutations are related because DNA damages often cause errors of DNA synthesis during replication or repair and these errors are a major source of mutation.
Given these properties of DNA damage and mutation, it can be seen that DNA damages are a special problem in non-dividing or slowly dividing cells, where unrepaired damages will tend to accumulate over time. On the other hand, in rapidly dividing cells, unrepaired DNA damages that do not kill the cell by blocking replication will tend to cause replication errors and thus mutation. The great majority of mutations that are not neutral in their effect are deleterious to a cell's survival. Thus, in a population of cells comprising a tissue with replicating cells, mutant cells will tend to be lost. However, infrequent mutations that provide a survival advantage will tend to clonally expand at the expense of neighboring cells in the tissue. This advantage to the cell is disadvantageous to the whole organism, because such mutant cells can give rise to cancer. Thus, DNA damages in frequently dividing cells, because they give rise to mutations, are a prominent cause of cancer. In contrast, DNA damages in infrequently dividing cells are likely a prominent cause of aging.
The first person to suggest that DNA damage, as distinct from mutation, is the primary cause of aging was Alexander in 1967.[14] By the early 1980s there was significant experimental support for this idea in the literature.[15] By the early 1990s experimental support for this idea was substantial, and furthermore it had become increasingly evident that oxidative DNA damage, in particular, is a major cause of aging.[16][17][13][18][19]
In a series of articles from 1970 to 1977, PV Narasimh Acharya, Phd. (1924–1993) theorized and presented evidence that cells undergo "irreparable DNA damage", whereby DNA crosslinks occur when both normal cellular repair processes fail and cellular apoptosis does not occur. Specifically, Acharya noted that double-strand breaks and a "cross-linkage joining both strands at the same point is irreparable because neither strand can then serve as a template for repair. The cell will die in the next mitosis or in some rare instances, mutate."[20][21][22][23][24]
Age-associated accumulation of DNA damage and changes in gene expression
In tissues composed of non- or infrequently replicating cells, DNA damage can accumulate with age and lead either to loss of cells, or, in surviving cells, loss of gene expression. Accumulated DNA damage is usually measured directly. Numerous studies of this type have indicated that oxidative damage to DNA is particularly important.[25] The loss of expression of specific genes can be detected at both the mRNA level and protein level.
Other form of age-associated changes in gene expression is increased transcriptional variability, that was found first in a selected panel of genes in heart cells [26] and, more recently, in the whole transcriptomes of immune cells,[27] and human pancreas cells.[28]
The adult brain is composed in large part of terminally differentiated non-dividing neurons. Many of the conspicuous features of aging reflect a decline in neuronal function. Accumulation of DNA damage with age in the mammalian brain has been reported during the period 1971 to 2008 in at least 29 studies.[29] This DNA damage includes the oxidized nucleoside 8-oxo-2'-deoxyguanosine (8-oxo-dG), single- and double-strand breaks, DNA-protein crosslinks and malondialdehyde adducts (reviewed in Bernstein et al.[29]). Increasing DNA damage with age has been reported in the brains of the mouse, rat, gerbil, rabbit, dog, and human.[13]
Rutten et al.[30] showed that single-strand breaks accumulate in the mouse brain with age. Young 4-day-old rats have about 3,000 single-strand breaks and 156 double-strand breaks per neuron, whereas in rats older than 2 years the level of damage increases to about 7,400 single-strand breaks and 600 double-strand breaks per neuron.[31] Sen et al.[32] showed that DNA damages which block the polymerase chain reaction in rat brain accumulate with age. Swain and Rao observed marked increases in several types of DNA damages in aging rat brain, including single-strand breaks, double-strand breaks and modified bases (8-OHdG and uracil).[33] Wolf et al.[34] also showed that the oxidative DNA damage 8-OHdG accumulates in rat brain with age. Similarly, it was shown that as humans age from 48 to 97 years, 8-OHdG accumulates in the brain.[35]
Lu et al.[36] studied the transcriptional profiles of the human frontal cortex of individuals ranging from 26 to 106 years of age. This led to the identification of a set of genes whose expression was altered after age 40. These genes play central roles in synaptic plasticity, vesicular transport and mitochondrial function. In the brain, promoters of genes with reduced expression have markedly increased DNA damage.[36] In cultured human neurons, these gene promoters are selectively damaged by oxidative stress. Thus Lu et al.[36] concluded that DNA damage may reduce the expression of selectively vulnerable genes involved in learning, memory and neuronal survival, initiating a program of brain aging that starts early in adult life.
Muscle strength, and stamina for sustained physical effort, decline in function with age in humans and other species. Skeletal muscle is a tissue composed largely of multinucleated myofibers, elements that arise from the fusion of mononucleated myoblasts. Accumulation of DNA damage with age in mammalian muscle has been reported in at least 18 studies since 1971.[29] Hamilton et al.[37] reported that the oxidative DNA damage 8-OHdG accumulates in heart and skeletal muscle (as well as in brain, kidney and liver) of both mouse and rat with age. In humans, increases in 8-OHdG with age were reported for skeletal muscle.[38] Catalase is an enzyme that removes hydrogen peroxide, a reactive oxygen species, and thus limits oxidative DNA damage. In mice, when catalase expression is increased specifically in mitochondria, oxidative DNA damage (8-OHdG) in skeletal muscle is decreased and lifespan is increased by about 20%.[39][40] These findings suggest that mitochondria are a significant source of the oxidative damages contributing to aging.
Protein synthesis and protein degradation decline with age in skeletal and heart muscle, as would be expected, since DNA damage blocks gene transcription. In 2005, Piec et al.[41] found numerous changes in protein expression in rat skeletal muscle with age, including lower levels of several proteins related to myosin and actin. Force is generated in striated muscle by the interactions between myosin thick filaments and actin thin filaments.
Liver hepatocytes do not ordinarily divide and appear to be terminally differentiated, but they retain the ability to proliferate when injured. With age, the mass of the liver decreases, blood flow is reduced, metabolism is impaired, and alterations in microcirculation occur. At least 21 studies have reported an increase in DNA damage with age in liver.[29] For instance, Helbock et al.[42] estimated that the steady state level of oxidative DNA base alterations increased from 24,000 per cell in the liver of young rats to 66,000 per cell in the liver of old rats.
One or two months after inducing DNA double-strand breaks in the livers of young mice, the mice showed multiple symptoms of aging similar to those seen in untreated livers of normally aged control mice.[43]
In kidney, changes with age include reduction in both renal blood flow and glomerular filtration rate, and impairment in the ability to concentrate urine and to conserve sodium and water. DNA damages, particularly oxidative DNA damages, increase with age (at least 8 studies).[29] For instance Hashimoto et al.[44] showed that 8-OHdG accumulates in rat kidney DNA with age.
Tissue-specific stem cells produce differentiated cells through a series of increasingly more committed progenitor intermediates. In hematopoiesis (blood cell formation), the process begins with long-term hematopoietic stem cells that self-renew and also produce progeny cells that upon further replication go through a series of stages leading to differentiated cells without self-renewal capacity. In mice, deficiencies in DNA repair appear to limit the capacity of hematopoietic stem cells to proliferate and self-renew with age.[45] Sharpless and Depinho reviewed evidence that hematopoietic stem cells, as well as stem cells in other tissues, undergo intrinsic aging.[46] They speculated that stem cells grow old, in part, as a result of DNA damage. DNA damage may trigger signalling pathways, such as apoptosis, that contribute to depletion of stem cell stocks. This has been observed in several cases of accelerated aging and may occur in normal aging too.[2]
A key aspect of hair loss with age is the aging of the hair follicle.[47] Ordinarily, hair follicle renewal is maintained by the stem cells associated with each follicle. Aging of the hair follicle appears to be due to the DNA damage that accumulates in renewing stem cells during aging.[48]
A related theory is that mutation, as distinct from DNA damage, is the primary cause of aging. A comparison of somatic mutation rate across several mammal species found that the total number of accumulated mutations at the end of lifespan was roughly equal across a broad range of lifespans.[49] The authors state that this strong relationship between somatic mutation rate and lifespan across different mammalian species suggests that evolution may constrain somatic mutation rates, perhaps by selection acting on different DNA repair pathways.[citation needed]
As discussed above, mutations tend to arise in frequently replicating cells as a result of errors of DNA synthesis when template DNA is damaged, and can give rise to cancer. However, in mice there is no increase in mutation in the brain with aging.[50][51][52] Mice defective in a gene (Pms2) that ordinarily corrects base mispairs in DNA have about a 100-fold elevated mutation frequency in all tissues, but do not appear to age more rapidly.[53] On the other hand, mice defective in one particular DNA repair pathway show clear premature aging, but do not have elevated mutation.[54]
One variation of the idea that mutation is the basis of aging, that has received much attention, is that mutations specifically in mitochondrial DNA are the cause of aging. Several studies have shown that mutations accumulate in mitochondrial DNA in infrequently replicating cells with age. DNA polymerase gamma is the enzyme that replicates mitochondrial DNA. A mouse mutant with a defect in this DNA polymerase is only able to replicate its mitochondrial DNA inaccurately, so that it sustains a 500-fold higher mutation burden than normal mice. These mice showed no clear features of rapidly accelerated aging.[55] Overall, the observations discussed in this section indicate that mutations are not the primary cause of aging.
In rodents, caloric restriction slows aging and extends lifespan. At least 4 studies have shown that caloric restriction reduces 8-OHdG damages in various organs of rodents. One of these studies showed that caloric restriction reduced accumulation of 8-OHdG with age in rat brain, heart and skeletal muscle, and in mouse brain, heart, kidney and liver.[37] More recently, Wolf et al.[34] showed that dietary restriction reduced accumulation of 8-OHdG with age in rat brain, heart, skeletal muscle, and liver. Thus reduction of oxidative DNA damage is associated with a slower rate of aging and increased lifespan.
If DNA damage is the underlying cause of aging, it would be expected that humans with inherited defects in the ability to repair DNA damages should age at a faster pace than persons without such a defect. Numerous examples of rare inherited conditions with DNA repair defects are known. Several of these show multiple striking features of premature aging, and others have fewer such features. Perhaps the most striking premature aging conditions are Werner syndrome (mean lifespan 47 years), Huchinson–Gilford progeria (mean lifespan 13 years), and Cockayne syndrome (mean lifespan 13 years).
Werner syndrome is due to an inherited defect in an enzyme (a helicase and exonuclease) that acts in base excision repair of DNA (e.g. see Harrigan et al.[56]).
Huchinson–Gilford progeria is due to a defect in Lamin A protein which forms a scaffolding within the cell nucleus to organize chromatin and is needed for repair of double-strand breaks in DNA.[57] A-type lamins promote genetic stability by maintaining levels of proteins that have key roles in the DNA repair processes of non-homologous end joining and homologous recombination.[58] Mouse cells deficient for maturation of prelamin A show increased DNA damage and chromosome aberrations and are more sensitive to DNA damaging agents.[59]
Cockayne Syndrome is due to a defect in a protein necessary for the repair process, transcription coupled nucleotide excision repair, which can remove damages, particularly oxidative DNA damages, that block transcription.[60]
In addition to human inherited syndromes, experimental mouse models with genetic defects in DNA repair show features of premature aging and reduced lifespan.(e.g. refs.[61][62][63]) In particular, mutant mice defective in Ku70, or Ku80, or double mutant mice deficient in both Ku70 and Ku80 exhibit early aging.[64] The mean lifespans of the three mutant mouse strains were similar to each other, at about 37 weeks, compared to 108 weeks for the wild-type control. Six specific signs of aging were examined, and the three mutant mice were found to display the same aging signs as the control mice, but at a much earlier age. Cancer incidence was not increased in the mutant mice. Ku70 and Ku80 form the heterodimer Ku protein essential for the non-homologous end joining (NHEJ) pathway of DNA repair, active in repairing DNA double-strand breaks. This suggests an important role of NHEJ in longevity assurance.
Defects in DNA repair cause features of premature aging
Many authors have noted an association between defects in the DNA damage response and premature aging (see e.g.[65][66][67][68]). If a DNA repair protein is deficient, unrepaired DNA damages tend to accumulate.[69] Such accumulated DNA damages appear to cause features of premature aging (segmental progeria). Table 1 lists 18 DNA repair proteins which, when deficient, cause numerous features of premature aging.
Table 1. DNA repair proteins that, when deficient, cause features of accelerated aging (segmental progeria).
deletion of ATR in adult mice leads to a number of disorders including hair loss and graying, kyphosis, osteoporosis, premature involution of the thymus, fibrosis of the heart and kidney and decreased spermatogenesis[66]
deficient transcription coupled NER with time-dependent accumulation of transcription-blocking damages;[75] mouse life span reduced from 2.5 years to 5 months;[68]Ercc1−/− mice are leukopenic and thrombocytopenic, and there is extensive adipose transformation of the bone marrow, hallmark features of normal aging in mice[74]
some mutations in ERCC2 cause Cockayne syndrome in which patients have segmental progeria with reduced stature, mental retardation, cachexia (loss of subcutaneous fat tissue), sensorineural deafness, retinal degeneration, and calcification of the central nervous system; other mutations in ERCC2 cause trichothiodystrophy in which patients have segmental progeria with brittle hair, short stature, progressive cognitive impairment and abnormal face shape; still other mutations in ERCC2 cause xeroderma pigmentosum (without a progeroid syndrome) and with extreme sun-mediated skin cancer predisposition[76]
mutations in ERCC4 cause symptoms of accelerated aging that affect the neurologic, hepatobiliary, musculoskeletal, and hematopoietic systems, and cause an old, wizened appearance, loss of subcutaneous fat, liver dysfunction, vision and hearing loss, renal insufficiency, muscle wasting, osteopenia, kyphosis and cerebral atrophy[74]
mice with deficient ERCC5 show loss of subcutaneous fat, kyphosis, osteoporosis, retinal photoreceptor loss, liver aging, extensive neurodegeneration, and a short lifespan of 4–5 months
premature aging features with shorter life span and photosensitivity,[81] deficient transcription coupled NER with accumulation of unrepaired DNA damages,[82] also defective repair of oxidatively generated DNA damages including 8-oxoguanine, 5-hydroxycytosine and cyclopurines[82]
premature aging features with shorter life span and photosensitivity,[81] deficient transcription coupled NER with accumulation of unrepaired DNA damages,[82] also defective repair of oxidatively generated DNA damages including 8-oxoguanine, 5-hydroxycytosine and cyclopurines[82]
deficiency causes trichothiodystrophy (TTD) a premature-ageing and neuroectodermal disease; humans with GTF2H5 mutations have a partially inactivated protein[83] with retarded repair of 6-4-photoproducts[84]
SIRT6-deficient mice develop profound lymphopenia, loss of subcutaneous fat and lordokyphosis, and these defects overlap with aging-associated degenerative processes[63]
mice defective in SIRT7 show phenotypic and molecular signs of accelerated aging such as premature pronounced curvature of the spine, reduced life span, and reduced non-homologous end joining[90]
lack of Zmpste24 prevents lamin A formation and causes progeroid phenotypes in mice and humans, increased DNA damage and chromosome aberrations, sensitivity to DNA-damaging agents and deficiency in homologous recombination[59]
Increased DNA repair and extended longevity
Table 2 lists DNA repair proteins whose increased expression is connected to extended longevity.
Table 2. DNA repair proteins that, when highly- or over-expressed, cause (or are associated with) extended longevity.
long-lived Snell dwarf, GHRKO, and PAPPA-KO mice have increased expression of NDRG1; higher expression of NDRG1 can promote MGMT protein stability and enhanced DNA repair[99][100]
degrades 8-oxodGTP; prevents the age-dependent accumulation of DNA 8-oxoguanine[101] A transgenic mouse in which the human hMTH1 8-oxodGTPase is expressed,[102] giving over-expression of hMTH1, increases the median lifespan of mice to 914 days vs. 790 days for wild-type mice.[101] Mice with over-expressed hMTH1 have behavioral changes of reduced anxiety and enhanced investigation of environmental and social cues
PARP1 activity in blood cells of thirteen mammalian species (rat, guinea pig, rabbit, marmoset, sheep, pig, cattle, pigmy chimpanzee, horse, donkey, gorilla, elephant and man) correlates with maximum lifespan of the species.[107]
Increased expression of SIRT1 in male mice extends the lifespan of mice fed a standard diet, accompanied by improvements in health, including enhanced motor coordination, performance, bone mineral density, and insulin sensitivity[109][110]
Studies comparing DNA repair capacity in different mammalian species have shown that repair capacity correlates with lifespan. The initial study of this type, by Hart and Setlow,[112] showed that the ability of skin fibroblasts of seven mammalian species to perform DNA repair after exposure to a DNA damaging agent correlated with lifespan of the species. The species studied were shrew, mouse, rat, hamster, cow, elephant and human. This initial study stimulated many additional studies involving a wide variety of mammalian species, and the correlation between repair capacity and lifespan generally held up. In one of the more recent studies, Burkle et al.[113] studied the level of a particular enzyme, Poly ADP ribose polymerase, which is involved in repair of single-strand breaks in DNA. They found that the lifespan of 13 mammalian species correlated with the activity of this enzyme.
The DNA repair transcriptomes of the liver of humans, naked mole-rats and mice were compared.[114] The maximum lifespans of humans, naked mole-rat, and mouse are respectively ~120, 30 and 3 years. The longer-lived species, humans and naked mole rats expressed DNA repair genes, including core genes in several DNA repair pathways, at a higher level than did mice. In addition, several DNA repair pathways in humans and naked mole-rats were up-regulated compared with mouse. These findings suggest that increased DNA repair facilitates greater longevity.
Over the past decade, a series of papers have shown that the mitochondrial DNA (mtDNA) base composition correlates with animal species maximum life span.[115][116][117][118] The mitochondrial DNA base composition is thought to reflect its nucleotide-specific (guanine, cytosine, thymidine and adenine) different mutation rates (i.e., accumulation of guanine in the mitochondrial DNA of an animal species is due to low guanine mutation rate in the mitochondria of that species).
DNA damage accumulation and repair decline
The rate of accumulation of DNA damage (double-strand breaks) in the leukocytes of dolphins, goats, reindeer, American flamingos, and griffon vultures was compared to the longevity of individuals of these different species.[119] The species with longer lifespans were found to have slower accumulation of DNA damage, a finding consistent with the DNA damage theory of aging.[119] In healthy humans after age 50, endogenous DNA single- and double-strand breaks increase linearly, and other forms of DNA damage also increase with age in blood mononuclear cells.[120] Also, after age 50 DNA repair capability decreases with age.[120]
In mice, the DNA repair process of non-homologous end-joining that repairs DNA double strand breaks, declines in efficiency from 1.8-3.8-fold, depending on the specific tissue, when 5 month old animals are compared to 24 month old animals.[121] A study of fibroblast cells from humans varying in age from 16-75 years showed that the efficiency and fidelity of non-homologous end joining, and the efficiency of homologous recombinational DNA repair decline with age leading to increased sensitivity to ionizing radiation in older individuals.[122] In middle aged human adults, oxidative DNA damage was found to be greater among individuals who were both frail and living in poverty.[123]
Lymphoblastoid cell lines established from blood samples of humans who lived past 100 years (centenarians) have significantly higher activity of the DNA repair protein Poly (ADP-ribose) polymerase (PARP) than cell lines from younger individuals (20 to 70 years old).[124][unreliable medical source?] The lymphocytic cells of centenarians have characteristics typical of cells from young people, both in their capability of priming the mechanism of repair after H2O2 sublethal oxidative DNA damage and in their PARP capacity.[10][125]
As women age, they experience a decline in reproductive performance leading to menopause. This decline is tied to a decline in the number of ovarian follicles. Although 6 to 7 million oocytes are present at mid-gestation in the human ovary,[127] only about 500 (about 0.05%) of these ovulate, and the rest are lost. The decline in ovarian reserve appears to occur at an increasing rate with age,[128][127] and leads to nearly complete exhaustion of the reserve by about age 51. As ovarian reserve and fertility decline with age, there is also a parallel increase in pregnancy failure and meiotic errors resulting in chromosomally abnormal conceptions.
BRCA1 and BRCA2 are homologous recombination repair genes. The role of declining ATM-Mediated DNA double strand DNA break (DSB) repair in oocyte aging was first proposed by Kutluk Oktay, MD, PhD based on his observations that women with BRCA mutations produced fewer oocytes in response to ovarian stimulation repair.[129][130][131] His laboratory has further studied this hypothesis and provided an explanation for the decline in ovarian reserve with age.[132] They showed that as women age, double-strand breaks accumulate in the DNA of their primordial follicles. Primordial follicles are immature primary oocytes surrounded by a single layer of granulosa cells. An enzyme system is present in oocytes that normally accurately repairs DNA double-strand breaks. This repair system is referred to as homologous recombinational repair, and it is especially active during meiosis. Titus et al.[132] from Oktay Laboratory also showed that expression of four key DNA repair genes that are necessary for homologous recombinational repair (BRCA1, MRE11, Rad51 and ATM) decline in oocytes with age. This age-related decline in ability to repair double-strand damages can account for the accumulation of these damages, which then likely contributes to the decline in ovarian reserve as further explained by Turan and Oktay.[133]
Women with an inherited mutation in the DNA repair gene BRCA1 undergo menopause prematurely,[134] suggesting that naturally occurring DNA damages in oocytes are repaired less efficiently in these women, and this inefficiency leads to early reproductive failure. Genomic data from about 70,000 women were analyzed to identify protein-coding variation associated with age at natural menopause.[135] Pathway analyses identified a major association with DNA damage response genes, particularly those expressed during meiosis and including a common coding variant in the BRCA1 gene.
Atherosclerosis
The most important risk factor for cardiovascular problems is chronological aging. Several research groups have reviewed evidence for a key role of DNA damage in vascular aging.[136][137][138]
Atherosclerotic plaque contains vascular smooth muscle cells, macrophages and endothelial cells and these have been found to accumulate 8-oxoG, a common type of oxidative DNA damage.[139] DNA strand breaks also increased in atherosclerotic plaques, thus linking DNA damage to plaque formation.[139]
Werner syndrome (WS), a premature aging condition in humans, is caused by a genetic defect in a RecQ helicase that is employed in several DNA repair processes. WS patients develop a substantial burden of atherosclerotic plaques in their coronary arteries and aorta.[137] These findings link excessive unrepaired DNA damage to premature aging and early atherosclerotic plaque development.
Several reviews[140][141][142] summarize evidence that the methylation enzyme DNMT1 is recruited to sites of oxidative DNA damage. Recruitment of DNMT1 leads to DNA methylation at the promoters of genes to inhibit transcription during repair. In addition, the 2018 review[140] describes recruitment of DNMT1 during repair of DNA double-strand breaks. DNMT1 localization results in increased DNA methylation near the site of recombinational repair, associated with altered expression of the repaired gene. In general, repair-associated hyper-methylated promoters are restored to their former methylation level after DNA repair is complete. However, these reviews also indicate that transient recruitment of epigenetic modifiers can occasionally result in subsequent stable epigenetic alterations and gene silencing after DNA repair has been completed.
In human and mouse DNA, cytosine followed by guanine (CpG) is the least frequent dinucleotide, making up less than 1% of all dinucleotides (see CG suppression). At most CpG sites cytosine is methylated to form 5-methylcytosine. As indicated in the article CpG site, in mammals, 70% to 80% of CpG cytosines are methylated. However, in vertebrates there are CpG islands, about 300 to 3,000 base pairs long, with interspersed DNA sequences that deviate significantly from the average genomic pattern by being CpG-rich. These CpG islands are predominantly nonmethylated.[143] In humans, about 70% of promoters located near the transcription start site of a gene (proximal promoters) contain a CpG island (see CpG islands in promoters). If the initially nonmethylated CpG sites in a CpG island become largely methylated, this causes stable silencing of the associated gene.
For humans, after adulthood is reached and during subsequent aging, the majority of CpG sequences slowly lose methylation (called epigenetic drift). However, the CpG islands that control promoters tend to gain methylation with age.[144] The gain of methylation at CpG islands in promoter regions is correlated with age, and has been used to create an epigenetic clock (see article Epigenetic clock).
There may be some relationship between the epigenetic clock and epigenetic alterations accumulating after DNA repair. Both unrepaired DNA damage accumulated with age and accumulated methylation of CpG islands would silence genes in which they occur, interfere with protein expression, and contribute to the aging phenotype.
DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur. This can eventually lead to malignant tumors, or cancer as per the two-hit hypothesis.
RecQ helicase is a family of helicase enzymes initially found in Escherichia coli that has been shown to be important in genome maintenance. They function through catalyzing the reaction ATP + H2O → ADP + P and thus driving the unwinding of paired DNA and translocating in the 3' to 5' direction. These enzymes can also drive the reaction NTP + H2O → NDP + P to drive the unwinding of either DNA or RNA.
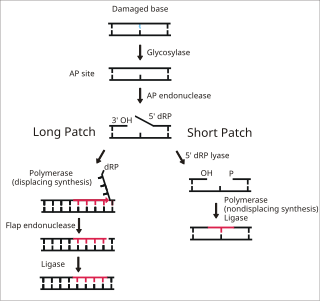
DNA glycosylases are a family of enzymes involved in base excision repair, classified under EC number EC 3.2.2. Base excision repair is the mechanism by which damaged bases in DNA are removed and replaced. DNA glycosylases catalyze the first step of this process. They remove the damaged nitrogenous base while leaving the sugar-phosphate backbone intact, creating an apurinic/apyrimidinic site, commonly referred to as an AP site. This is accomplished by flipping the damaged base out of the double helix followed by cleavage of the N-glycosidic bond.
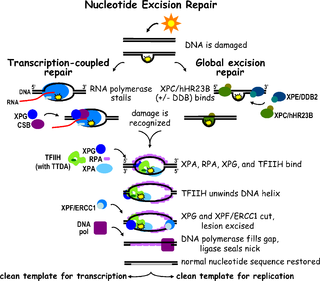
Nucleotide excision repair is a DNA repair mechanism. DNA damage occurs constantly because of chemicals, radiation and other mutagens. Three excision repair pathways exist to repair single stranded DNA damage: Nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR). While the BER pathway can recognize specific non-bulky lesions in DNA, it can correct only damaged bases that are removed by specific glycosylases. Similarly, the MMR pathway only targets mismatched Watson-Crick base pairs.
Base excision repair (BER) is a cellular mechanism, studied in the fields of biochemistry and genetics, that repairs damaged DNA throughout the cell cycle. It is responsible primarily for removing small, non-helix-distorting base lesions from the genome. The related nucleotide excision repair pathway repairs bulky helix-distorting lesions. BER is important for removing damaged bases that could otherwise cause mutations by mispairing or lead to breaks in DNA during replication. BER is initiated by DNA glycosylases, which recognize and remove specific damaged or inappropriate bases, forming AP sites. These are then cleaved by an AP endonuclease. The resulting single-strand break can then be processed by either short-patch or long-patch BER.
MUTYH is a human gene that encodes a DNA glycosylase, MUTYH glycosylase. It is involved in oxidative DNA damage repair and is part of the base excision repair pathway. The enzyme excises adenine bases from the DNA backbone at sites where adenine is inappropriately paired with guanine, cytosine, or 8-oxo-7,8-dihydroguanine, a common form of oxidative DNA damage.
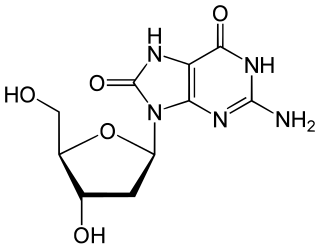
DNA oxidation is the process of oxidative damage of deoxyribonucleic acid. As described in detail by Burrows et al., 8-oxo-2'-deoxyguanosine (8-oxo-dG) is the most common oxidative lesion observed in duplex DNA because guanine has a lower one-electron reduction potential than the other nucleosides in DNA. The one electron reduction potentials of the nucleosides are guanine 1.29, adenine 1.42, cytosine 1.6 and thymine 1.7. About 1 in 40,000 guanines in the genome are present as 8-oxo-dG under normal conditions. This means that >30,000 8-oxo-dGs may exist at any given time in the genome of a human cell. Another product of DNA oxidation is 8-oxo-dA. 8-oxo-dA occurs at about 1/10 the frequency of 8-oxo-dG. The reduction potential of guanine may be reduced by as much as 50%, depending on the particular neighboring nucleosides stacked next to it within DNA.
Cell damage is a variety of changes of stress that a cell suffers due to external as well as internal environmental changes. Amongst other causes, this can be due to physical, chemical, infectious, biological, nutritional or immunological factors. Cell damage can be reversible or irreversible. Depending on the extent of injury, the cellular response may be adaptive and where possible, homeostasis is restored. Cell death occurs when the severity of the injury exceeds the cell's ability to repair itself. Cell death is relative to both the length of exposure to a harmful stimulus and the severity of the damage caused. Cell death may occur by necrosis or apoptosis.
A DNA repair-deficiency disorder is a medical condition due to reduced functionality of DNA repair.
TFIIH subunit XPD is a protein that in humans is encoded by the ERCC2 gene. It is a component of the general transcription and DNA repair factor IIH (TFIIH) core complex involved in transcription-coupled nucleotide excision repair.
DNA repair protein XRCC1, also known as X-ray repair cross-complementing protein 1, is a protein that in humans is encoded by the XRCC1 gene. XRCC1 is involved in DNA repair, where it complexes with DNA ligase III.
8-Oxoguanine glycosylase, also known as OGG1, is a DNA glycosylase enzyme that, in humans, is encoded by the OGG1 gene. It is involved in base excision repair. It is found in bacterial, archaeal and eukaryotic species.
DNA excision repair protein ERCC-1 is a protein that in humans is encoded by the ERCC1 gene. Together with ERCC4, ERCC1 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.
DNA polymerase beta, also known as POLB, is an enzyme present in eukaryotes. In humans, it is encoded by the POLB gene.
DNA excision repair protein ERCC-6 is a protein that in humans is encoded by the ERCC6 gene. The ERCC6 gene is located on the long arm of chromosome 10 at position 11.23.
DNA repair protein complementing XP-G cells is a protein that in humans is encoded by the ERCC5 gene.
8-Oxo-2'-deoxyguanosine (8-oxo-dG) is an oxidized derivative of deoxyguanosine. 8-Oxo-dG is one of the major products of DNA oxidation. Concentrations of 8-oxo-dG within a cell are a measurement of oxidative stress.
Genome instability refers to a high frequency of mutations within the genome of a cellular lineage. These mutations can include changes in nucleic acid sequences, chromosomal rearrangements or aneuploidy. Genome instability does occur in bacteria. In multicellular organisms genome instability is central to carcinogenesis, and in humans it is also a factor in some neurodegenerative diseases such as amyotrophic lateral sclerosis or the neuromuscular disease myotonic dystrophy.
DNA damage is an alteration in the chemical structure of DNA, such as a break in a strand of DNA, a nucleobase missing from the backbone of DNA, or a chemically changed base such as 8-OHdG. DNA damage can occur naturally or via environmental factors, but is distinctly different from mutation, although both are types of error in DNA. DNA damage is an abnormal chemical structure in DNA, while a mutation is a change in the sequence of base pairs. DNA damages cause changes in the structure of the genetic material and prevents the replication mechanism from functioning and performing properly. The DNA damage response (DDR) is a complex signal transduction pathway which recognizes when DNA is damaged and initiates the cellular response to the damage.
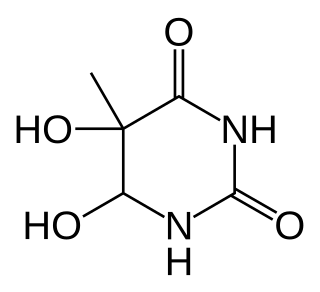
Thymine glycol (5,6-dihydroxy-5,6-dihydrothymine) is one of the principal DNA lesions that can be induced by oxidation and ionizing radiation.
1 2 3 Holmes GE, Bernstein C, Bernstein H (September 1992). "Oxidative and other DNA damages as the basis of aging: a review". Mutat Res. 275 (3–6): 305–15. doi:10.1016/0921-8734(92)90034-m. PMID1383772.
↑ Alexander, P. (1967). The role of DNA lesions in the processes leading to aging in mice. Symp Soc Exp Biol. Vol.21. pp.29–50. PMID4860956.
↑ Rao, K. S.; Loeb, L. A. (September 1992). "DNA damage and repair in brain: relationship to aging". Mutation Research/DNAging. 275 (3–6): 317–329. doi:10.1016/0921-8734(92)90035-N. PMID1383773.
↑ Acharya, P. V. (1972). "The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides". Johns Hopkins Med. J. Suppl. (1): 254–260. PMID5055816.
↑ Acharya, P. V.; Ashman, S. M.; Bjorksten, J (1972). "The isolation and partial characterization of age-correlated oligo-deoxyribo-ribo nucleo peptides". Finska Kemists Medd. 81 (3).
↑ Acharya, P. V. N. (June 19, 1971). Isolation and Partial Characterization of Age-Correlated Oligo-nucleotides with Covalently Bound Peptides. 14th Nordic Congress. Umeå, Sweden.
↑ Acharya, P. V. N. (July 1–7, 1973). DNA-damage: The Cause of Aging. Ninth International Congress of Biochemistry. Stockholm.
↑ Acharya, P. V. N. (1977). "Irreparable DNA-damage by Industrial Pollutants in Pre-mature Aging, Chemical Carcinogenesis and Cardiac Hypertrophy: Experiments and Theory". Israel Journal of Medical Sciences. 13: 441.
↑ Sinha, Jitendra Kumar; Ghosh, Shampa; Swain, Umakanta; Giridharan, Nappan Veethil; Raghunath, Manchala (2014). "Increased macromolecular damage due to oxidative stress in the neocortex and hippocampus of WNIN/Ob, a novel rat model of premature aging". Neuroscience. 269: 256–64. doi:10.1016/j.neuroscience.2014.03.040. PMID24709042. S2CID9934178.
↑ Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J (June 2006). "Increased cell-to-cell variation in gene expression in ageing mouse heart". Nature. 441 (7096): 1011–4. doi:10.1038/nature04844. PMID16791200.
1 2 3 4 5 Bernstein H, Payne CM, Bernstein C, Garewal H, Dvorak K (2008). "1. Cancer and aging as consequences of un-repaired DNA damage". In Kimura H, Suzuki A (eds.). New Research on DNA Damages. Nova Science. pp.1–47. ISBN978-1-60456-581-2. OCLC213848806.
↑ Rutten, BP; Schmitz, C; Gerlach, OH; Oyen, HM; de Mesquita, EB; Steinbusch, HW; Korr, H (Jan 2007). "The aging brain: accumulation of DNA damage or neuron loss?". Neurobiol Aging. 28 (1): 91–8. doi:10.1016/j.neurobiolaging.2005.10.019. PMID16338029. S2CID14620944.
↑ Sen, T; Jana, S; Sreetama, S; Chatterjee, U; Chakrabarti, S (Mar 2007). "Gene-specific oxidative lesions in aged rat brain detected by polymerase chain reaction inhibition assay". Free Radic. Res. 41 (3): 288–94. doi:10.1080/10715760601083722. PMID17364957. S2CID23610941.
↑ Swain, U; Subba Rao, K (Aug 2011). "Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging". Mech Ageing Dev. 132 (8–9): 374–81. doi:10.1016/j.mad.2011.04.012. PMID21600238. S2CID22466782.
1 2 Wolf, FI; Fasanella, S; Tedesco, B; Cavallini, G; Donati, A; Bergamini, E; Cittadini, A (Mar 2005). "Peripheral lymphocyte 8-OHdG levels correlate with age-associated increase of tissue oxidative DNA damage in Sprague-Dawley rats. Protective effects of caloric restriction". Exp Gerontol. 40 (3): 181–8. doi:10.1016/j.exger.2004.11.002. PMID15763395. S2CID23752647.
↑ Mecocci, P; MacGarvey, U; Kaufman, AE; Koontz, D; Shoffner, JM; Wallace, DC; Beal, MF (Oct 1993). "Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain". Ann Neurol. 34 (4): 609–16. doi:10.1002/ana.410340416. PMID8215249. S2CID25479410.
↑ Mecocci, P.; Fanó, G.; Fulle, S.; MacGarvey, U.; Shinobu, L.; Polidori, M. C.; Cherubini, A; Vecchiet, J.; Senin, U.; Beal, M. F. (February 1999). "Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle". Free Radic Biol Med. 26 (3–4): 303–8. doi:10.1016/s0891-5849(98)00208-1. PMID9895220.
↑ Schriner, S. E.; Linford, NJ; Martin, G. M.; Treuting, P.; Ogburn, C. E.; Emond, M.; Coskun, P. E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; Wallace, D. C.; Rabinovitch, P. S. (June 2005). "Extension of murine life span by overexpression of catalase targeted to mitochondria". Science. 308 (5730): 1909–11. Bibcode:2005Sci...308.1909S. doi:10.1126/science.1106653. PMID15879174. S2CID38568666.
↑ Sharpless, NE; DePinho, RA (Sep 2007). "How stem cells age and why this makes us grow old". Nat Rev Mol Cell Biol. 8 (9): 703–13. doi:10.1038/nrm2241. PMID17717515. S2CID36305591.
↑ Matsumura H, Mohri Y, Binh NT, Morinaga H, Fukuda M, Ito M, Kurata S, Hoeijmakers J, Nishimura EK (2016). "Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis". Science. 351 (6273): aad4395. doi:10.1126/science.aad4395. PMID26912707. S2CID5078019.
↑ Hill, KA; Halangoda, A; Heinmoeller, PW; Gonzalez, K; Chitaphan, C; Longmate, J; Scaringe, WA; Wang, JC; Sommer, SS (Jun 2005). "Tissue-specific time courses of spontaneous mutation frequency and deviations in mutation pattern are observed in middle to late adulthood in Big Blue mice". Environ Mol Mutagen. 45 (5): 442–54. Bibcode:2005EnvMM..45..442H. doi:10.1002/em.20119. PMID15690342. S2CID32204458.
↑ Dollé, ME; Busuttil, RA; Garcia, AM; Wijnhoven, S; van Drunen, E; Niedernhofer, LJ; van der Horst, G; Hoeijmakers, JH; van Steeg, H; Vijg, J (Apr 2006). "Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice". Mutat. Res. 596 (1–2): 22–35. Bibcode:2006MRFMM.596...22D. doi:10.1016/j.mrfmmm.2005.11.008. PMID16472827.
↑ Vermulst, M; Bielas, JH; Kujoth, GC; Ladiges, WC; Rabinovitch, PS; Prolla, TA; Loeb, LA (Apr 2007). "Mitochondrial point mutations do not limit the natural lifespan of mice". Nat Genet. 39 (4): 540–3. doi:10.1038/ng1988. PMID17334366. S2CID291780.
↑ Weinfeld M, Xing JZ, Lee J, Leadon SA, Cooper PK, Le XC (2001). "Factors influencing the removal of thymine glycol from DNA in γ-irradiated human cells". Base Excision Repair. Progress in Nucleic Acid Research and Molecular Biology. Vol.68. pp.139–49. doi:10.1016/S0079-6603(01)68096-6. ISBN978-0-12-540068-8. PMID11554293.
↑ Bürkle, A; Brabeck, C; Diefenbach, J; Beneke, S (May 2005). "The emerging role of poly(ADP-ribose) polymerase-1 in longevity". Int J Biochem Cell Biol. 37 (5): 1043–53. doi:10.1016/j.biocel.2004.10.006. PMID15743677.
↑ Lehmann, Gilad; Budovsky, Arie; Muradian, K. Muradian; Fraifeld, Vadim E. (2006). "Mitochondrial genome anatomy and species-specific lifespan". Rejuvenation Res. 9 (2): 223–6. doi:10.1089/rej.2006.9.223. PMID16706648.
↑ Lehmann, Gilad; Segal, Elena; Muradian, K. Muradian; Fraifeld, Vadim E. (2008). "Do mitochondrial DNA and metabolic rate complement each other in determination of the mammalian maximum longevity?". Rejuvenation Res. 11 (2): 409–417. doi:10.1089/rej.2008.0676. PMID18442324.
↑ Rzepka-Górska I, Tarnowski B, Chudecka-Głaz A, Górski B, Zielińska D, Tołoczko-Grabarek A (2006). "Premature menopause in patients with BRCA1 gene mutation". Breast Cancer Res. Treat. 100 (1): 59–63. doi:10.1007/s10549-006-9220-1. PMID16773440. S2CID19572648.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.