Related Research Articles

Analytical chemistry studies and uses instruments and methods to separate, identify, and quantify matter. In practice, separation, identification or quantification may constitute the entire analysis or be combined with another method. Separation isolates analytes. Qualitative analysis identifies analytes, while quantitative analysis determines the numerical amount or concentration.

Atomic absorption spectroscopy (AAS) and atomic emission spectroscopy (AES) is a spectroanalytical procedure for the quantitative determination of chemical elements by free atoms in the gaseous state. Atomic absorption spectroscopy is based on absorption of light by free metallic ions.

Spectrophotometry is a branch of electromagnetic spectroscopy concerned with the quantitative measurement of the reflection or transmission properties of a material as a function of wavelength. Spectrophotometry uses photometers, known as spectrophotometers, that can measure the intensity of a light beam at different wavelengths. Although spectrophotometry is most commonly applied to ultraviolet, visible, and infrared radiation, modern spectrophotometers can interrogate wide swaths of the electromagnetic spectrum, including x-ray, ultraviolet, visible, infrared, and/or microwave wavelengths.
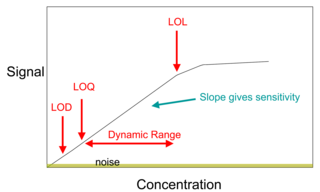
In analytical chemistry, a calibration curve, also known as a standard curve, is a general method for determining the concentration of a substance in an unknown sample by comparing the unknown to a set of standard samples of known concentration. A calibration curve is one approach to the problem of instrument calibration; other standard approaches may mix the standard into the unknown, giving an internal standard. The calibration curve is a plot of how the instrumental response, the so-called analytical signal, changes with the concentration of the analyte.
Analytical technique is a method used to determine a chemical or physical property of a chemical substance, chemical element, or mixture. There is a wide variety of techniques used for analysis, from simple weighing to advanced techniques using highly specialized instrumentation.

Near-infrared spectroscopy (NIRS) is a spectroscopic method that uses the near-infrared region of the electromagnetic spectrum. Typical applications include medical and physiological diagnostics and research including blood sugar, pulse oximetry, functional neuroimaging, sports medicine, elite sports training, ergonomics, rehabilitation, neonatal research, brain computer interface, urology, and neurology. There are also applications in other areas as well such as pharmaceutical, food and agrochemical quality control, atmospheric chemistry, combustion research and knowledge.

Metabolomics is the scientific study of chemical processes involving metabolites, the small molecule substrates, intermediates, and products of cell metabolism. Specifically, metabolomics is the "systematic study of the unique chemical fingerprints that specific cellular processes leave behind", the study of their small-molecule metabolite profiles. The metabolome represents the complete set of metabolites in a biological cell, tissue, organ, or organism, which are the end products of cellular processes. Messenger RNA (mRNA), gene expression data, and proteomic analyses reveal the set of gene products being produced in the cell, data that represents one aspect of cellular function. Conversely, metabolic profiling can give an instantaneous snapshot of the physiology of that cell, and thus, metabolomics provides a direct "functional readout of the physiological state" of an organism. There are indeed quantifiable correlations between the metabolome and the other cellular ensembles, which can be used to predict metabolite abundances in biological samples from, for example mRNA abundances. One of the ultimate challenges of systems biology is to integrate metabolomics with all other -omics information to provide a better understanding of cellular biology.
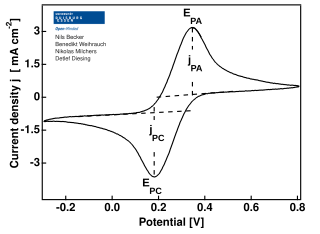
In electrochemistry, cyclic voltammetry (CV) is a type of potentiodynamic measurement. In a cyclic voltammetry experiment, the working electrode potential is ramped linearly versus time. Unlike in linear sweep voltammetry, after the set potential is reached in a CV experiment, the working electrode's potential is ramped in the opposite direction to return to the initial potential. These cycles of ramps in potential may be repeated as many times as needed. The current at the working electrode is plotted versus the applied voltage to give the cyclic voltammogram trace. Cyclic voltammetry is generally used to study the electrochemical properties of an analyte in solution or of a molecule that is adsorbed onto the electrode.
The limit of detection is the lowest signal, or the lowest corresponding quantity to be determined from the signal, that can be observed with a sufficient degree of confidence or statistical significance. However, the exact threshold used to decide when a signal significantly emerges above the continuously fluctuating background noise remains arbitrary and is a matter of policy and often of debate among scientists, statisticians and regulators depending on the stakes in different fields.
In a chemical analysis, the internal standard method involves adding the same amount of a chemical substance to each sample and calibration solution. The internal standard responds proportionally to changes in the analyte and provides a similar, but not identical, measurement signal. It must also be absent from the sample matrix to ensure there is no other source of the internal standard present. Taking the ratio of analyte signal to internal standard signal and plotting it against the analyte concentrations in the calibration solutions will result in a calibration curve. The calibration curve can then be used to calculate the analyte concentration in an unknown sample.
Chemical imaging is the analytical capability to create a visual image of components distribution from simultaneous measurement of spectra and spatial, time information. Hyperspectral imaging measures contiguous spectral bands, as opposed to multispectral imaging which measures spaced spectral bands.
Multivariate optical computing, also known as molecular factor computing, is an approach to the development of compressed sensing spectroscopic instruments, particularly for industrial applications such as process analytical support. "Conventional" spectroscopic methods often employ multivariate and chemometric methods, such as multivariate calibration, pattern recognition, and classification, to extract analytical information from data collected at many different wavelengths. Multivariate optical computing uses an optical computer to analyze the data as it is collected. The goal of this approach is to produce instruments which are simple and rugged, yet retain the benefits of multivariate techniques for the accuracy and precision of the result.

The Journal of Chemometrics is a monthly peer-reviewed scientific journal published since 1987 by John Wiley & Sons. It publishes original scientific papers, reviews, and short communications on fundamental and applied aspects of chemometrics. The current editor-in-chief is Age K. Smilde.
Flow injection analysis (FIA) is an approach to chemical analysis. It is accomplished by injecting a plug of sample into a flowing carrier stream. The principle is similar to that of Segmented Flow Analysis (SFA) but no air is injected into the sample or reagent streams..
The Unscrambler X is a commercial software product for multivariate data analysis, used for calibration of multivariate data which is often in the application of analytical data such as near infrared spectroscopy and Raman spectroscopy, and development of predictive models for use in real-time spectroscopic analysis of materials. The software was originally developed in 1986 by Harald Martens and later by CAMO Software.
Acoustic resonance spectroscopy (ARS) is a method of spectroscopy in the acoustic region, primarily the sonic and ultrasonic regions. ARS is typically much more rapid than HPLC and NIR. It is non destructive and requires no sample preparation as the sampling waveguide can simply be pushed into a sample powder/liquid or in contact with a solid sample.
Paul J. Gemperline is an American analytical chemist and chemometrician. He is a Distinguished Professor of Chemistry at East Carolina University (ECU) located in Greenville, North Carolina and has been the recipient of several scientific awards, including the 2003 Eastern Analytical Symposium Award in Chemometrics. He is author of more than 60 publications in the field of chemometrics. Dr. Gemperline served as Dean of the Graduate School at ECU from 2008 to 2022. He retired from ECU June 30, 2022 and is now professor emeritus.
Multiway data analysis is a method of analyzing large data sets by representing a collection of observations as a multiway array, . The proper choice of data organization into (C+1)-way array, and analysis techniques can reveal patterns in the underlying data undetected by other methods.
Bruce R. Kowalski was an American professor of analytical chemistry who is acknowledged by the world-wide scientific community to be one of the founders of the field of chemometrics. He was the founding editor of Journal of Chemometrics, and the founding director of the Center for Process Analytical Chemistry at University of Washington in Seattle. Kowalski and Svante Wold formed the Chemometrics Society, which would later become the International Chemometrics Society.

Tormod Næs is a Norwegian statistician working in chemometrics and sensometrics. He studies multivariate statistical analysis, spectroscopy, food science, and sensory science. His impact on chemometrics is exemplified by the over 8,000 citations to his most well-known book, Multivariate Calibration, and the awards in chemometrics that he has received.
References
- ↑ As recounted in Wold, S. (1995). "Chemometrics; what do we mean with it, and what do we want from it?". Chemometrics and Intelligent Laboratory Systems. 30 (1): 109–115. doi:10.1016/0169-7439(95)00042-9.
- ↑ Kowalski, Bruce R. (1975). "Chemometrics: Views and Propositions". J. Chem. Inf. Comput. Sci. 15 (4): 201–203. doi:10.1021/ci60004a002.
- ↑ Trygg, J.; Wold, S. (2003). "O2-PLS, a two-block (X–Y) latent variable regression (LVR) method with an integral OSC filter". Journal of Chemometrics. 17: 53–64. doi:10.1002/cem.775. S2CID 123071521.
- ↑ Malinowski, E. R.; Howery, D. G. (1980). Factor Analysis in Chemistry. New York: Wiley. ISBN 978-0471058816. (other editions followed in 1989, 1991 and 2002).
- ↑ Sharaf, M. A.; Illman, D. L.; Kowalski, B. R., eds. (1986). Chemometrics. New York: Wiley. ISBN 978-0471831068.
- ↑ Massart, D. L.; Vandeginste, B. G. M.; Deming, S. M.; Michotte, Y.; Kaufman, L. (1988). Chemometrics: a textbook. Amsterdam: Elsevier. ISBN 978-0444426604.
- 1 2 Martens, H.; Naes, T. (1989). Multivariate Calibration. New York: Wiley. ISBN 978-0471909798.
- ↑ Geladi, P.; Esbensen, K. (2005). "The Start and Early History of Chemometrics: Selected Interviews. Part 1". J. Chemometrics . 4 (5): 337–354. doi:10.1002/cem.1180040503. S2CID 120490459.
- ↑ Esbensen, K.; Geladi, P. (2005). "The Start and Early History of Chemometrics: Selected Interviews. Part 2". J. Chemometrics . 4 (6): 389–412. doi:10.1002/cem.1180040604. S2CID 221546473.
- ↑ Barton, Bastian; Thomson, James; Lozano Diz, Enrique; Portela, Raquel (September 2022). "Chemometrics for Raman Spectroscopy Harmonization". Applied Spectroscopy. 76 (9): 1021–1041. Bibcode:2022ApSpe..76.1021B. doi:10.1177/00037028221094070. ISSN 0003-7028. PMID 35622984. S2CID 249129065.
- ↑ Franke, J. (2002). "Inverse Least Squares and Classical Least Squares Methods for Quantitative Vibrational Spectroscopy". In Chalmers, John M (ed.). Handbook of Vibrational Spectroscopy. New York: Wiley. doi:10.1002/0470027320.s4603. ISBN 978-0471988472.
- ↑ Brown, C. D. (2004). "Discordance between Net Analyte Signal Theory and Practical Multivariate Calibration". Analytical Chemistry. 76 (15): 4364–4373. doi:10.1021/ac049953w. PMID 15283574.
- ↑ Krutchkoff, R. G. (1969). "Classical and inverse regression methods of calibration in extrapolation". Technometrics. 11 (3): 11–15. doi:10.1080/00401706.1969.10490714.
- ↑ Hunter, W. G. (1984). "Statistics and chemistry, and the linear calibration problem". In Kowalski, B. R. (ed.). Chemometrics: mathematics and statistics in chemistry. Boston: Riedel. ISBN 978-9027718464.
- ↑ Tellinghuisen, J. (2000). "Inverse vs. classical calibration for small data sets". Fresenius' J. Anal. Chem. 368 (6): 585–588. doi:10.1007/s002160000556. PMID 11228707. S2CID 21166415.
- ↑ Oliveri, Paolo (2017). "Class-modelling in food analytical chemistry: Development, sampling, optimisation and validation issues – A tutorial". Analytica Chimica Acta. 982: 9–19. Bibcode:2017AcAC..982....9O. doi:10.1016/j.aca.2017.05.013. hdl: 11567/881059 . PMID 28734370. S2CID 10119515.
- ↑ Lawton, W. H.; Sylvestre, E. A. (1971). "Self Modeling Curve Resolution". Technometrics. 13 (3): 617–633. doi:10.1080/00401706.1971.10488823.
- ↑ Sylvestre, E. A.; Lawton, W. H.; Maggio, M. S. (1974). "Curve Resolution Using a Postulated Chemical Reaction". Technometrics. 16 (3): 353–368. doi:10.1080/00401706.1974.10489204.
- ↑ de Juan, A.; Tauler, R. (2003). "Chemometrics Applied to Unravel Multicomponent Processes and Mixtures. Revisiting Latest Trends in Multivariate Resolution". Analytica Chimica Acta. 500 (1–2): 195–210. Bibcode:2003AcAC..500..195D. doi:10.1016/S0003-2670(03)00724-4.
- ↑ de Juan, A.; Tauler, R. (2006). "Multivariate Curve Resolution (MCR) from 2000: Progress in Concepts and Applications". Critical Reviews in Analytical Chemistry. 36 (3–4): 163–176. doi:10.1080/10408340600970005. S2CID 95309963.
- ↑ Deming, S. N.; Morgan, S. L. (1987). Experimental design: a chemometric approach. Elsevier. ISBN 978-0444427342.
- ↑ Bruns, R. E.; Scarminio, I. S.; de Barros Neto, B. (2006). Statistical design – chemometrics. Amsterdam: Elsevier. ISBN 978-0444521811.
- ↑ Wentzell, P. D.; Brown, C. D. (2000). "Signal Processing in Analytical Chemistry". In Meyers, R. A. (ed.). Encyclopedia of Analytical Chemistry. Wiley. pp. 9764–9800.
- ↑ Oliveri, Paolo; Malegori, Cristina; Simonetti, Remo; Casale, Monica (2019). "The impact of signal pre-processing on the final interpretation of analytical outcomes – A tutorial". Analytica Chimica Acta. 1058: 9–17. Bibcode:2019AcAC.1058....9O. doi:10.1016/j.aca.2018.10.055. PMID 30851858. S2CID 73727614.
- ↑ Olivieri, A. C.; Faber, N. M.; Ferre, J.; Boque, R.; Kalivas, J. H.; Mark, H. (2006). "Guidelines for calibration in analytical chemistry Part 3. Uncertainty estimation and figures of merit for multivariate calibration". Pure and Applied Chemistry. 78 (3): 633–650. doi: 10.1351/pac200678030633 . S2CID 50546210.
- ↑ Illman, D. L.; Callis, J. B.; Kowalski, B. R. (1986). "Process Analytical Chemistry: a new paradigm for analytical chemists". American Laboratory. 18: 8–10.
- ↑ MacGregor, J. F.; Kourti, T. (1995). "Statistical control of multivariate processes". Control Engineering Practice. 3 (3): 403–414. doi:10.1016/0967-0661(95)00014-L.
- ↑ Martin, E. B.; Morris, A. J. (1996). "An overview of multivariate statistical process control in continuous and batch process performance monitoring". Transactions of the Institute of Measurement & Control. 18 (1): 51–60. Bibcode:1996TIMC...18...51M. doi:10.1177/014233129601800107. S2CID 120516715.
- ↑ Hirschfeld, T.; Callis, J. B.; Kowalski, B. R. (1984). "Chemical sensing in process analysis". Science . 226 (4672): 312–318. Bibcode:1984Sci...226..312H. doi:10.1126/science.226.4672.312. PMID 17749872. S2CID 38093353.
- ↑ Smilde, A. K.; Bro, R.; Geladi, P. (2004). Multi-way analysis with applications in the chemical sciences. Wiley.
- ↑ Bro, R.; Workman, J. J.; Mobley, P. R.; Kowalski, B. R. (1997). "Overview of chemometrics applied to spectroscopy: 1985–95, Part 3—Multiway analysis". Applied Spectroscopy Reviews. 32 (3): 237–261. Bibcode:1997ApSRv..32..237B. doi:10.1080/05704929708003315.