
Opioid receptors are a group of inhibitory G protein-coupled receptors with opioids as ligands. The endogenous opioids are dynorphins, enkephalins, endorphins, endomorphins and nociceptin. The opioid receptors are ~40% identical to somatostatin receptors (SSTRs). Opioid receptors are distributed widely in the brain, in the spinal cord, on peripheral neurons, and digestive tract.

Nalbuphine, sold under the brand names Nubain among others, is an opioid analgesic which is used in the treatment of pain. It is given by injection into a vein, muscle, or fat.

Dihydromorphine is a semi-synthetic opioid structurally related to and derived from morphine. The 7,8-double bond in morphine is reduced to a single bond to get dihydromorphine. Dihydromorphine is a moderately strong analgesic and is used clinically in the treatment of pain and also is an active metabolite of the analgesic opioid drug dihydrocodeine. Dihydromorphine occurs in trace quantities in assays of opium on occasion, as does dihydrocodeine, dihydrothebaine, tetrahydrothebaine, etc. The process for manufacturing dihydromorphine from morphine for pharmaceutical use was developed in Germany in the late 19th century, with the synthesis being published in 1900 and the drug introduced clinically as Paramorfan shortly thereafter. A high-yield synthesis from tetrahydrothebaine was later developed.
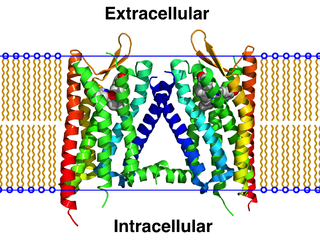
The κ-opioid receptor or kappa opioid receptor, abbreviated KOR or KOP for its ligand ketazocine, is a G protein-coupled receptor that in humans is encoded by the OPRK1 gene. The KOR is coupled to the G protein Gi/G0 and is one of four related receptors that bind opioid-like compounds in the brain and are responsible for mediating the effects of these compounds. These effects include altering nociception, consciousness, motor control, and mood. Dysregulation of this receptor system has been implicated in alcohol and drug addiction.

The nociceptin opioid peptide receptor (NOP), also known as the nociceptin/orphanin FQ (N/OFQ) receptor or kappa-type 3 opioid receptor, is a protein that in humans is encoded by the OPRL1 gene. The nociceptin receptor is a member of the opioid subfamily of G protein-coupled receptors whose natural ligand is the 17 amino acid neuropeptide known as nociceptin (N/OFQ). This receptor is involved in the regulation of numerous brain activities, particularly instinctive and emotional behaviors. Antagonists targeting NOP are under investigation for their role as treatments for depression and Parkinson's disease, whereas NOP agonists have been shown to act as powerful, non-addictive painkillers in non-human primates.
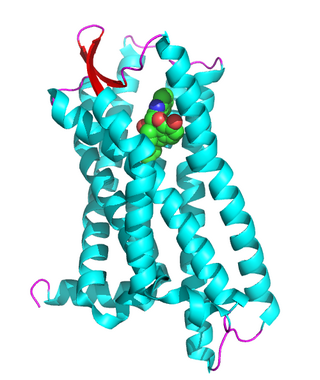
The δ-opioid receptor, also known as delta opioid receptor or simply delta receptor, abbreviated DOR or DOP, is an inhibitory 7-transmembrane G-protein coupled receptor coupled to the G protein Gi/G0 and has enkephalins as its endogenous ligands. The regions of the brain where the δ-opioid receptor is largely expressed vary from species model to species model. In humans, the δ-opioid receptor is most heavily expressed in the basal ganglia and neocortical regions of the brain.

SNC-80 is an opioid analgesic compound that selectively activates μ–δ opioid receptor heteromers and is used primarily in scientific research. Discovered in 1994, SNC-80 was a pioneering non-peptide compound regarded as a highly selective agonist for the δ-opioid receptor.

Proglumide, sold under the brand name Milid, is a drug that inhibits gastrointestinal motility and reduces gastric secretions. It acts as a cholecystokinin antagonist, which blocks both the CCKA and CCKB subtypes. It was used mainly in the treatment of stomach ulcers, although it has now been largely replaced by newer drugs for this application.
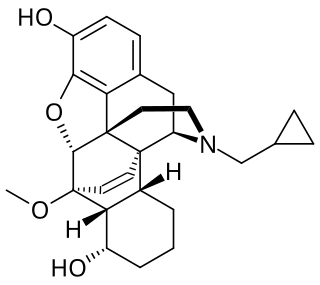
BU-48 is a drug that is used in scientific research. It is from the oripavine family, related to better-known drugs such as etorphine and buprenorphine.

DPI-221 is an opioid drug that is used in scientific research. It is a highly selective agonist for the δ-opioid receptor, which produces fewer convulsions than most drugs from this family.

DPI-287 is an opioid drug that is used in scientific research. It is a highly selective agonist for the δ-opioid receptor, which produces less convulsions than most drugs from this family. It has antidepressant-like effects.
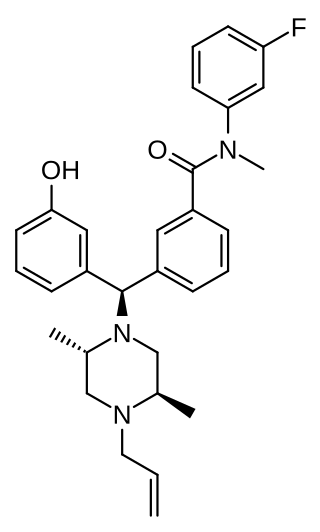
DPI-3290 was discovered by scientists at Burroughs Wellcome and licensed to Delta Pharmaceutical and is a drug that is used in scientific research. It is a potent analgesic drug, which produces little respiratory depression.

Xorphanol (INN), also known as xorphanol mesylate (USAN), is an opioid analgesic of the morphinan family that was never marketed.
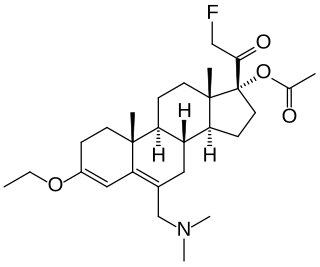
SC-17599 is a steroid derivative drug discovered in 1968 which acts as a selective μ-opioid receptor agonist, with little or no affinity for the δ-opioid or κ-opioid receptors. It is an active analgesic in vivo, more potent than codeine or pethidine but slightly less potent than morphine, and produces similar effects to morphine in animals but with less sedation

In pharmacology the term agonist-antagonist or mixed agonist/antagonist is used to refer to a drug which under some conditions behaves as an agonist while under other conditions, behaves as an antagonist.

TAN-67 (SB-205,607) is an opioid drug used in scientific research that acts as a potent and selective δ-opioid agonist, selective for the δ1 subtype. It has analgesic properties and induces dopamine release in nucleus accumbens. It also protects both heart and brain tissue from hypoxic tissue damage through multiple mechanisms involving among others an interaction between δ receptors and mitochondrial K(ATP) channels.
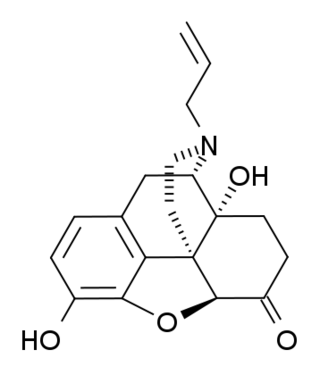
(+)-Naloxone (dextro-naloxone) is a drug which is the opposite enantiomer of the opioid antagonist drug (−)-naloxone. Unlike (−)-naloxone, (+)-naloxone has no significant affinity for opioid receptors, but instead has been discovered to act as a selective antagonist of Toll-like receptor 4. This receptor is involved in immune system responses, and activation of TLR4 induces glial activation and release of inflammatory mediators such as TNF-α and Interleukin-1.

SoRI-9409 is a mixed mu opioid receptor partial agonist and delta opioid receptor antagonist, used in biomedical research. It produces moderate analgesic effects without development of tolerance and with reduced withdrawal symptoms compared to standard opioid analgesics, as well as showing anti-addictive effects that may be useful in the treatment of alcoholism.

PZM21 is an experimental opioid analgesic drug that is being researched for the treatment of pain. It is claimed to be a functionally selective μ-opioid receptor agonist which produces μ-opioid receptor mediated G protein signaling, with potency and efficacy similar to morphine, but with less β-arrestin 2 recruitment. However, recent reports highlight that this might be due to its low intrinsic efficacy, rather than functional selectivity or 'G protein bias' as initially reported. In tests on mice, PZM21 was slightly less potent than morphine or TRV130 as an analgesic, but also had significantly reduced adverse effects, with less constipation than morphine, and very little respiratory depression, even at high doses. This research was described as a compelling example of how modern high-throughput screening techniques can be used to discover new chemotypes with specific activity profiles, even at targets such as the μ-opioid receptor which have already been thoroughly investigated. More recent research has suggested however that at higher doses, PZM21 is capable of producing classic opioid side effects such as respiratory depression and development of tolerance and may have only limited functional selectivity.

β-Funaltrexamine (β-FNA) is an irreversible opioid antagonist that was used to create the first crystal structure of the μ-opioid receptor (MOR). It is selective for antagonism of the MOR over the δ-opioid receptor (DOR) and κ-opioid receptor (KOR). Chemically, it is a naltrexone derivative with a methyl-fumaramide group in the 6-position. In addition to its MOR irreversible antagonism, β-FNA is a reversible agonist of the κ-opioid receptor (KOR) and produces KOR-mediated analgesic effects in animals. This has limited its usefulness and contributed to the development of methocinnamox as a more selective functionally irreversible antagonist of the MOR with no significant opioid agonistic actions.
