
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosome abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.
Barth syndrome (BTHS) is a rare but serious X-linked genetic disorder, caused by changes in phospholipid structure and metabolism. It may affect multiple body systems, and is potentially fatal. The syndrome is diagnosed almost exclusively in males.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Uniparental disomy (UPD) occurs when a person receives two copies of a chromosome, or of part of a chromosome, from one parent and no copy from the other. UPD can be the result of heterodisomy, in which a pair of non-identical chromosomes are inherited from one parent or isodisomy, in which a single chromosome from one parent is duplicated. Uniparental disomy may have clinical relevance for several reasons. For example, either isodisomy or heterodisomy can disrupt parent-specific genomic imprinting, resulting in imprinting disorders. Additionally, isodisomy leads to large blocks of homozygosity, which may lead to the uncovering of recessive genes, a similar phenomenon seen in inbred children of consanguineous partners.

Alport syndrome is a genetic disorder affecting around 1 in 5,000–10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

X-linked recessive inheritance is a mode of inheritance in which a mutation in a gene on the X chromosome causes the phenotype to be always expressed in males and in females who are homozygous for the gene mutation, see zygosity. Females with one copy of the mutated gene are carriers.

Sex linked describes the sex-specific reading patterns of inheritance and presentation when a gene mutation (allele) is present on a sex chromosome (allosome) rather than a non-sex chromosome (autosome). In humans, these are termed X-linked recessive, X-linked dominant and Y-linked. The inheritance and presentation of all three differ depending on the sex of both the parent and the child. This makes them characteristically different from autosomal dominance and recessiveness.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.
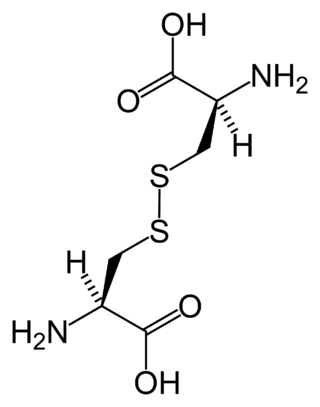
Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.
A lipid storage disorder is any one of a group of inherited metabolic disorders in which harmful amounts of fats or lipids accumulate in some body cells and tissues. People with these disorders either do not produce enough of one of the enzymes needed to metabolize and break down lipids or, they produce enzymes that do not work properly. Over time, the buildup of fats may cause permanent cellular and tissue damage, particularly in the brain, peripheral nervous system, liver, spleen, and bone marrow.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.
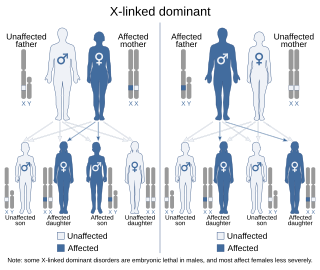
X-linked dominant inheritance, sometimes referred to as X-linked dominance, is a mode of genetic inheritance by which a dominant gene is carried on the X chromosome. As an inheritance pattern, it is less common than the X-linked recessive type. In medicine, X-linked dominant inheritance indicates that a gene responsible for a genetic disorder is located on the X chromosome, and only one copy of the allele is sufficient to cause the disorder when inherited from a parent who has the disorder. In this case, someone who expresses an X-linked dominant allele will exhibit the disorder and be considered affected. The pattern of inheritance is sometimes called criss-cross inheritance.

Costeff syndrome, or 3-methylglutaconic aciduria type III, is a genetic disorder caused by mutations in the OPA3 gene. It is typically associated with the onset of visual deterioration in early childhood followed by the development of movement problems and motor disability in later childhood, occasionally along with mild cases of cognitive deficiency. The disorder is named after Hanan Costeff, the doctor who first described the syndrome in 1989.

Hypermethioninemia is an excess of the amino acid methionine, in the blood. This condition can occur when methionine is not broken down properly in the body.
Pseudodominance is the situation in which the inheritance of a recessive trait mimics a dominant pattern.

Optic atrophy 3 protein is a protein that in humans is encoded by the OPA3 gene.
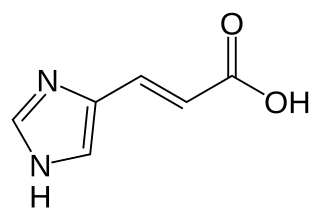
Urocanic aciduria is an autosomal recessive metabolic disorder caused by a deficiency of the enzyme urocanase. It is a secondary disorder of histidine metabolism.

Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.

Mohr–Tranebjærg syndrome (MTS) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. It is characterized by clinical manifestations commencing with early childhood onset hearing loss, followed by adolescent onset progressive or , visual impairment from early adulthood onwards and dementia from the 4th decade onwards. The severity of the symptoms may vary, but they progress usually to severe deafness and dystonia and sometimes are accompanied by cortical deterioration of vision and mental deterioration.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.
