
A dendritic spine is a small membrane protrusion from a neuron's dendrite that typically receives input from a single axon at the synapse. Dendritic spines serve as a storage site for synaptic strength and help transmit electrical signals to the neuron's cell body. Most spines have a bulbous head, and a thin neck that connects the head of the spine to the shaft of the dendrite. The dendrites of a single neuron can contain hundreds to thousands of spines. In addition to spines providing an anatomical substrate for memory storage and synaptic transmission, they may also serve to increase the number of possible contacts between neurons. It has also been suggested that changes in the activity of neurons have a positive effect on spine morphology.
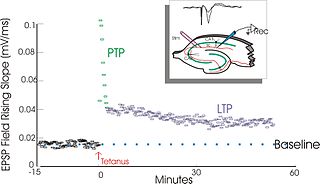
In neuroscience, long-term potentiation (LTP) is a persistent strengthening of synapses based on recent patterns of activity. These are patterns of synaptic activity that produce a long-lasting increase in signal transmission between two neurons. The opposite of LTP is long-term depression, which produces a long-lasting decrease in synaptic strength.

The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (also known as AMPA receptor, AMPAR, or quisqualate receptor) is an ionotropic transmembrane receptor for glutamate (iGluR) and predominantly Na+ ion channel that mediates fast synaptic transmission in the central nervous system (CNS). It has been traditionally classified as a non-NMDA-type receptor, along with the kainate receptor. Its name is derived from its ability to be activated by the artificial glutamate analog AMPA. The receptor was first named the "quisqualate receptor" by Watkins and colleagues after a naturally occurring agonist quisqualate and was only later given the label "AMPA receptor" after the selective agonist developed by Tage Honore and colleagues at the Royal Danish School of Pharmacy in Copenhagen. The GRIA2-encoded AMPA receptor ligand binding core (GluA2 LBD) was the first glutamate receptor ion channel domain to be crystallized.
In neurophysiology, long-term depression (LTD) is an activity-dependent reduction in the efficacy of neuronal synapses lasting hours or longer following a long patterned stimulus. LTD occurs in many areas of the CNS with varying mechanisms depending upon brain region and developmental progress.
In neuroscience, a silent synapse is an excitatory glutamatergic synapse whose postsynaptic membrane contains NMDA-type glutamate receptors but no AMPA-type glutamate receptors. These synapses are named "silent" because normal AMPA receptor-mediated signaling is not present, rendering the synapse inactive under typical conditions. Silent synapses are typically considered to be immature glutamatergic synapses. As the brain matures, the relative number of silent synapses decreases. However, recent research on hippocampal silent synapses shows that while they may indeed be a developmental landmark in the formation of a synapse, that synapses can be "silenced" by activity, even once they have acquired AMPA receptors. Thus, silence may be a state that synapses can visit many times during their lifetimes.
Spike-timing-dependent plasticity (STDP) is a biological process that adjusts the strength of connections between neurons in the brain. The process adjusts the connection strengths based on the relative timing of a particular neuron's output and input action potentials. The STDP process partially explains the activity-dependent development of nervous systems, especially with regard to long-term potentiation and long-term depression.
Schaffer collaterals are axon collaterals given off by CA3 pyramidal cells in the hippocampus. These collaterals project to area CA1 of the hippocampus and are an integral part of memory formation and the emotional network of the Papez circuit, and of the hippocampal trisynaptic loop. It is one of the most studied synapses in the world and named after the Hungarian anatomist-neurologist Károly Schaffer.
Metaplasticity is a term originally coined by W.C. Abraham and M.F. Bear to refer to the plasticity of synaptic plasticity. Until that time synaptic plasticity had referred to the plastic nature of individual synapses. However this new form referred to the plasticity of the plasticity itself, thus the term meta-plasticity. The idea is that the synapse's previous history of activity determines its current plasticity. This may play a role in some of the underlying mechanisms thought to be important in memory and learning such as long-term potentiation (LTP), long-term depression (LTD) and so forth. These mechanisms depend on current synaptic "state", as set by ongoing extrinsic influences such as the level of synaptic inhibition, the activity of modulatory afferents such as catecholamines, and the pool of hormones affecting the synapses under study. Recently, it has become clear that the prior history of synaptic activity is an additional variable that influences the synaptic state, and thereby the degree, of LTP or LTD produced by a given experimental protocol. In a sense, then, synaptic plasticity is governed by an activity-dependent plasticity of the synaptic state; such plasticity of synaptic plasticity has been termed metaplasticity. There is little known about metaplasticity, and there is much research currently underway on the subject, despite its difficulty of study, because of its theoretical importance in brain and cognitive science. Most research of this type is done via cultured hippocampus cells or hippocampal slices.
Ca2+
/calmodulin-dependent protein kinase II is a serine/threonine-specific protein kinase that is regulated by the Ca2+
/calmodulin complex. CaMKII is involved in many signaling cascades and is thought to be an important mediator of learning and memory. CaMKII is also necessary for Ca2+
homeostasis and reuptake in cardiomyocytes, chloride transport in epithelia, positive T-cell selection, and CD8 T-cell activation.
In neuroscience, homeostatic plasticity refers to the capacity of neurons to regulate their own excitability relative to network activity. The term homeostatic plasticity derives from two opposing concepts: 'homeostatic' and plasticity, thus homeostatic plasticity means "staying the same through change". In the nervous system, neurons must be able to evolve with the development of their constantly changing environment while simultaneously staying the same amidst this change. This stability is important for neurons to maintain their activity and functionality to prevent neurons from carcinogenesis. At the same time, neurons need to have flexibility to adapt to changes and make connections to cope with the ever-changing environment of a developing nervous system.
Coincidence detection is a neuronal process in which a neural circuit encodes information by detecting the occurrence of temporally close but spatially distributed input signals. Coincidence detectors influence neuronal information processing by reducing temporal jitter and spontaneous activity, allowing the creation of variable associations between separate neural events in memory. The study of coincidence detectors has been crucial in neuroscience with regards to understanding the formation of computational maps in the brain.
The spine apparatus (SA) is a specialized form of endoplasmic reticulum (ER) that is found in a subpopulation of dendritic spines in central neurons. It was discovered by Edward George Gray in 1959 when he applied electron microscopy to fixed cortical tissue. The SA consists of a series of stacked discs that are connected to each other and to the dendritic system of ER-tubules. The actin binding protein synaptopodin is an essential component of the SA. Mice that lack the gene for synaptopodin do not form a spine apparatus. The SA is believed to play a role in synaptic plasticity, learning and memory, but the exact function of the spine apparatus is still enigmatic.
Activity-dependent plasticity is a form of functional and structural neuroplasticity that arises from the use of cognitive functions and personal experience. Hence, it is the biological basis for learning and the formation of new memories. Activity-dependent plasticity is a form of neuroplasticity that arises from intrinsic or endogenous activity, as opposed to forms of neuroplasticity that arise from extrinsic or exogenous factors, such as electrical brain stimulation- or drug-induced neuroplasticity. The brain's ability to remodel itself forms the basis of the brain's capacity to retain memories, improve motor function, and enhance comprehension and speech amongst other things. It is this trait to retain and form memories that is associated with neural plasticity and therefore many of the functions individuals perform on a daily basis. This plasticity occurs as a result of changes in gene expression which are triggered by signaling cascades that are activated by various signaling molecules during increased neuronal activity.

Nonsynaptic plasticity is a form of neuroplasticity that involves modification of ion channel function in the axon, dendrites, and cell body that results in specific changes in the integration of excitatory postsynaptic potentials and inhibitory postsynaptic potentials. Nonsynaptic plasticity is a modification of the intrinsic excitability of the neuron. It interacts with synaptic plasticity, but it is considered a separate entity from synaptic plasticity. Intrinsic modification of the electrical properties of neurons plays a role in many aspects of plasticity from homeostatic plasticity to learning and memory itself. Nonsynaptic plasticity affects synaptic integration, subthreshold propagation, spike generation, and other fundamental mechanisms of neurons at the cellular level. These individual neuronal alterations can result in changes in higher brain function, especially learning and memory. However, as an emerging field in neuroscience, much of the knowledge about nonsynaptic plasticity is uncertain and still requires further investigation to better define its role in brain function and behavior.
Synaptic tagging, or the synaptic tagging hypothesis, has been proposed to explain how neural signaling at a particular synapse creates a target for subsequent plasticity-related product (PRP) trafficking essential for sustained LTP and LTD. Although the molecular identity of the tags remains unknown, it has been established that they form as a result of high or low frequency stimulation, interact with incoming PRPs, and have a limited lifespan.
In neuroscience, synaptic scaling is a form of homeostatic plasticity, in which the brain responds to chronically elevated activity in a neural circuit with negative feedback, allowing individual neurons to reduce their overall action potential firing rate. Where Hebbian plasticity mechanisms modify neural synaptic connections selectively, synaptic scaling normalizes all neural synaptic connections by decreasing the strength of each synapse by the same factor, so that the relative synaptic weighting of each synapse is preserved.
Long-term potentiation (LTP), thought to be the cellular basis for learning and memory, involves a specific signal transmission process that underlies synaptic plasticity. Among the many mechanisms responsible for the maintenance of synaptic plasticity is the cadherin–catenin complex. By forming complexes with intracellular catenin proteins, neural cadherins (N-cadherins) serve as a link between synaptic activity and synaptic plasticity, and play important roles in the processes of learning and memory.
Addiction is a state characterized by compulsive engagement in rewarding stimuli, despite adverse consequences. The process of developing an addiction occurs through instrumental learning, which is otherwise known as operant conditioning.
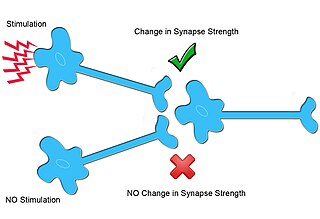
Homosynaptic plasticity is one type of synaptic plasticity. Homosynaptic plasticity is input-specific, meaning changes in synapse strength occur only at post-synaptic targets specifically stimulated by a pre-synaptic target. Therefore, the spread of the signal from the pre-synaptic cell is localized.

Synaptic stabilization is crucial in the developing and adult nervous systems and is considered a result of the late phase of long-term potentiation (LTP). The mechanism involves strengthening and maintaining active synapses through increased expression of cytoskeletal and extracellular matrix elements and postsynaptic scaffold proteins, while pruning less active ones. For example, cell adhesion molecules (CAMs) play a large role in synaptic maintenance and stabilization. Gerald Edelman discovered CAMs and studied their function during development, which showed CAMs are required for cell migration and the formation of the entire nervous system. In the adult nervous system, CAMs play an integral role in synaptic plasticity relating to learning and memory.







