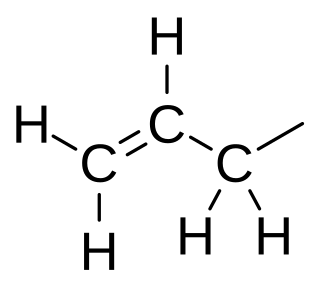
In organic chemistry, an allyl group is a substituent with the structural formula −CH2−HC=CH2. It consists of a methylene bridge attached to a vinyl group. The name is derived from the scientific name for garlic, Allium sativum. In 1844, Theodor Wertheim isolated an allyl derivative from garlic oil and named it "Schwefelallyl". The term allyl applies to many compounds related to H2C=CH−CH2, some of which are of practical or of everyday importance, for example, allyl chloride.
The Wolff–Kishner reduction is a reaction used in organic chemistry to convert carbonyl functionalities into methylene groups. In the context of complex molecule synthesis, it is most frequently employed to remove a carbonyl group after it has served its synthetic purpose of activating an intermediate in a preceding step. As such, there is no obvious retron for this reaction. The reaction was reported by Nikolai Kischner in 1911 and Ludwig Wolff in 1912.
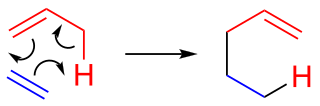
In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.
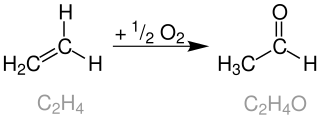
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide called a Wittig reagent. Wittig reactions are most commonly used to convert aldehydes and ketones to alkenes. Most often, the Wittig reaction is used to introduce a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent, even a sterically hindered ketone such as camphor can be converted to its methylene derivative.
The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant. The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.
Organoselenium chemistry is the science exploring the properties and reactivity of organoselenium compounds, chemical compounds containing carbon-to-selenium chemical bonds. Selenium belongs with oxygen and sulfur to the group 16 elements or chalcogens, and similarities in chemistry are to be expected. Organoselenium compounds are found at trace levels in ambient waters, soils and sediments.
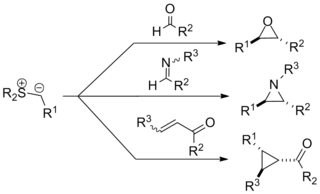
The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.
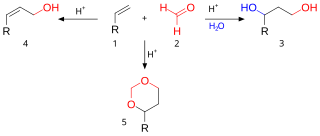
The Prins reaction is an organic reaction consisting of an electrophilic addition of an aldehyde or ketone to an alkene or alkyne followed by capture of a nucleophile or elimination of an H+ ion. The outcome of the reaction depends on reaction conditions. With water and a protic acid such as sulfuric acid as the reaction medium and formaldehyde the reaction product is a 1,3-diol (3). When water is absent, the cationic intermediate loses a proton to give an allylic alcohol (4). With an excess of formaldehyde and a low reaction temperature the reaction product is a dioxane (5). When water is replaced by acetic acid the corresponding esters are formed.
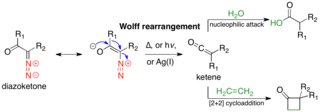
The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The Lemieux–Johnson or Malaprade–Lemieux–Johnson oxidation is a chemical reaction in which an olefin undergoes oxidative cleavage to form two aldehyde or ketone units. The reaction is named after its inventors, Raymond Urgel Lemieux and William Summer Johnson, who published it in 1956. The reaction proceeds in a two step manner, beginning with dihydroxylation of the alkene by osmium tetroxide, followed by a Malaprade reaction to cleave the diol using periodate. Periodate also serves to regenerate the osmium tetroxide. This means a only catalytic amount of the osmium reagent is needed and also that the two consecutive reactions can be performed as a single tandem reaction process. The Lemieux–Johnson reaction ceases at the aldehyde stage of oxidation and therefore produces the same results as ozonolysis.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.
Oxidation with chromium(VI) complexes involves the conversion of alcohols to carbonyl compounds or more highly oxidized products through the action of molecular chromium(VI) oxides and salts. The principal reagents are Collins reagent, PDC, and PCC. These reagents represent improvements over inorganic chromium(VI) reagents such as Jones reagent.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
The Saegusa–Ito oxidation is a chemical reaction used in organic chemistry. It was discovered in 1978 by Takeo Saegusa and Yoshihiko Ito as a method to introduce α-β unsaturation in carbonyl compounds. The reaction as originally reported involved formation of a silyl enol ether followed by treatment with palladium(II) acetate and benzoquinone to yield the corresponding enone. The original publication noted its utility for regeneration of unsaturation following 1,4-addition with nucleophiles such as organocuprates.
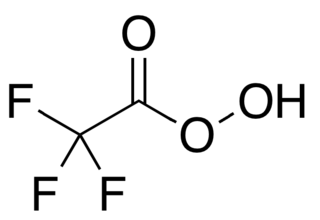
Trifluoroperacetic acid is an organofluorine compound, the peroxy acid analog of trifluoroacetic acid, with the condensed structural formula CF
3COOOH. It is a strong oxidizing agent for organic oxidation reactions, such as in Baeyer–Villiger oxidations of ketones. It is the most reactive of the organic peroxy acids, allowing it to successfully oxidise relatively unreactive alkenes to epoxides where other peroxy acids are ineffective. It can also oxidise the chalcogens in some functional groups, such as by transforming selenoethers to selones. It is a potentially explosive material and is not commercially available, but it can be quickly prepared as needed. Its use as a laboratory reagent was pioneered and developed by William D. Emmons.
Carbonyl olefin metathesis is a type of metathesis reaction that entails, formally, the redistribution of fragments of an alkene and a carbonyl by the scission and regeneration of carbon-carbon and carbon-oxygen double bonds respectively. It is a powerful method in organic synthesis using simple carbonyls and olefins and converting them into less accessible products with higher structural complexity.











