Periodic fever syndromes are a set of disorders characterized by recurrent episodes of systemic and organ-specific inflammation. Unlike autoimmune disorders such as systemic lupus erythematosus, in which the disease is caused by abnormalities of the adaptive immune system, people with autoinflammatory diseases do not produce autoantibodies or antigen-specific T or B cells. Instead, the autoinflammatory diseases are characterized by errors in the innate immune system.

TNF receptor associated periodic syndrome (TRAPS) is a periodic fever syndrome associated with mutations in a receptor for the molecule tumor necrosis factor (TNF) that is inheritable in an autosomal dominant manner. Individuals with TRAPS have episodic symptoms such as recurrent high fevers, rash, abdominal pain, joint/muscle aches and puffy eyes.
Lymphedema–distichiasis syndrome is a medical condition associated with the FOXC2 gene. People with this hereditary condition have a double row of eyelashes, which is called distichiasis, and a risk of swollen limbs due to problems in the lymphatic system.
Papillorenal syndrome is an autosomal dominant genetic disorder marked by underdevelopment (hypoplasia) of the kidney and colobomas of the optic nerve.

NF-kappa-B essential modulator (NEMO) also known as inhibitor of nuclear factor kappa-B kinase subunit gamma (IKK-γ) is a protein that in humans is encoded by the IKBKG gene. NEMO is a subunit of the IκB kinase complex that activates NF-κB. The human gene for IKBKG is located on the chromosome band Xq28. Multiple transcript variants encoding different isoforms have been found for this gene.

Signal transducer and activator of transcription 1 (STAT1) is a transcription factor which in humans is encoded by the STAT1 gene. It is a member of the STAT protein family.

Toll-like receptor 5, also known as TLR5, is a protein which in humans is encoded by the TLR5 gene. It is a member of the toll-like receptor (TLR) family. TLR5 is known to recognize bacterial flagellin from invading mobile bacteria. It has been shown to be involved in the onset of many diseases, including Inflammatory bowel disease due to the high expression of TLR in intestinal lamina propria dendritic cells. Recent studies have also shown that malfunctioning of TLR5 is likely related to rheumatoid arthritis, osteoclastogenesis, and bone loss. Abnormal TLR5 functioning is related to the onset of gastric, cervical, endometrial and ovarian cancers.

Caspase recruitment domain-containing protein 9 is an adaptor protein of the CARD-CC protein family, which in humans is encoded by the CARD9 gene. It mediates signals from pattern recognition receptors to activate pro-inflammatory and anti-inflammatory cytokines, regulating inflammation. Homozygous mutations in CARD9 are associated with defective innate immunity against yeasts, like Candida and dermatophytes.

The nucleotide-binding oligomerization domain-like receptors, or NOD-like receptors (NLRs), are intracellular sensors of pathogen-associated molecular patterns (PAMPs) that enter the cell via phagocytosis or pores, and damage-associated molecular patterns (DAMPs) that are associated with cell stress. They are types of pattern recognition receptors (PRRs), and play key roles in the regulation of innate immune response. NLRs can cooperate with toll-like receptors (TLRs) and regulate inflammatory and apoptotic response.
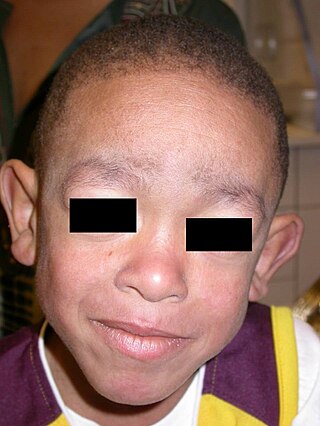
Blau syndrome is an autosomal dominant genetic inflammatory disorder which affects the skin, eyes, and joints. It is caused by a mutation in the NOD2 (CARD15) gene. and is classified as an inborn errors of immunity. Symptoms usually begin before the age of four, and the disease manifests as early onset cutaneous sarcoidosis, granulomatous arthritis, and uveitis.
MonoMAC syndrome is a rare autosomal dominant syndrome associated with: monocytopenia, B and NK cell lymphopenia; mycobacterial, viral, fungal, and bacterial opportunistic infections; and virus infection-induced cancers. The disorder often progresses to the development of myelodysplasia, myeloid leukemias, and other types of cancer. MonoMAC is a life-threatening and precancerous disorder.
BENTA disease is a rare genetic disorder of the immune system. BENTA stands for "B cell expansion with NF-κB and T cell anergy" and is caused by germline heterozygous gain-of-function mutations in the gene CARD11. This disorder is characterized by polyclonal B cell lymphocytosis with onset in infancy, splenomegaly, lymphadenopathy, mild immunodeficiency, and increased risk of lymphoma. Investigators Andrew L. Snow and Michael J. Lenardo at the National Institute of Allergy and Infectious Diseases at the U.S. National Institutes of Health first characterized BENTA disease in 2012. Dr. Snow's current laboratory at the Uniformed Services University of the Health Sciences is now actively studying this disorder.

Caspase-8 deficiency (CEDS) is a very rare genetic disorder of the immune system. It is caused by mutations in the CASP8 gene that encodes the protein caspase-8. The disorder is characterized by splenomegaly and lymphadenopathy, in addition to recurrent sinopulmonary infections, recurrent mucocutaneous herpesvirus or other viral infections, and hypogammaglobulinemia. Investigators in the laboratory of Dr. Michael Lenardo at the National Institutes of Health described this condition in two siblings from a consanguineous family in 2002, and several more affected family members have since been identified.
Activated PI3K delta syndrome (APDS) is a primary immunodeficiency disease caused by activating gain of function mutations in the PIK3CD gene.
Nuclear factor-kappa B Essential Modulator (NEMO) deficiency syndrome is a rare type of primary immunodeficiency disease that has a highly variable set of symptoms and prognoses. It mainly affects the skin and immune system but has the potential to affect all parts of the body, including the lungs, urinary tract and gastrointestinal tract. It is a monogenetic disease caused by mutation in the IKBKG gene. NEMO is the modulator protein in the IKK inhibitor complex that, when activated, phosphorylates the inhibitor of the NF-κB transcription factors allowing for the translocation of transcription factors into the nucleus.
Birk-Barel syndrome is a rare genetic disorder associated with the KCNK9 gene. Signs and symptoms include mental retardation, hypotonia, hyperactivity, and syndromic facies.
Autoinflammatory diseases (AIDs) are a group of rare disorders caused by dysfunction of the innate immune system. These responses are characterized by periodic or chronic systemic inflammation, usually without the involvement of adaptive immunity.

Hypohidrotic/anhidrotic ectodermal dysplasia with immune deficiency is a rare genetic condition characterized by a combination of the features of ectodermal dysplasia alongside immunodeficiency.
Wolfram-like syndrome is a rare autosomal dominant genetic disorder that shares some of the features shown by those affected with the autosomal recessive Wolfram syndrome. It is a type of WFS1-related disorder.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
