
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

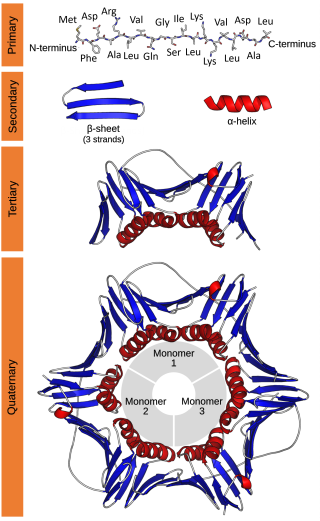
Protein primary structure is the linear sequence of amino acids in a peptide or protein. By convention, the primary structure of a protein is reported starting from the amino-terminal (N) end to the carboxyl-terminal (C) end. Protein biosynthesis is most commonly performed by ribosomes in cells. Peptides can also be synthesized in the laboratory. Protein primary structures can be directly sequenced, or inferred from DNA sequences.

Protein biosynthesis is a core biological process, occurring inside cells, balancing the loss of cellular proteins through the production of new proteins. Proteins perform a number of critical functions as enzymes, structural proteins or hormones. Protein synthesis is a very similar process for both prokaryotes and eukaryotes but there are some distinct differences.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

Protein folding is the physical process by which a protein, after synthesis by a ribosome as a linear chain of amino acids, changes from an unstable random coil into a more ordered three-dimensional structure. This structure permits the protein to become biologically functional.
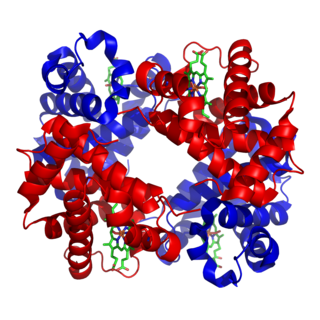
In biochemistry, globular proteins or spheroproteins are spherical ("globe-like") proteins and are one of the common protein types. Globular proteins are somewhat water-soluble, unlike the fibrous or membrane proteins. There are multiple fold classes of globular proteins, since there are many different architectures that can fold into a roughly spherical shape.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.
Levinthal's paradox is a thought experiment in the field of computational protein structure prediction; protein folding seeks a stable energy configuration. An algorithmic search through all possible conformations to identify the minimum energy configuration would take an immense duration, however, in reality, protein folding happens very quickly, even in the case of the most complex structures, suggesting that the transitions are guided into a stable state through an uneven energy landscape.
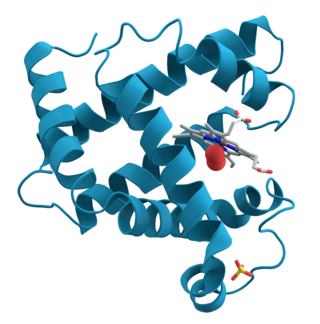
Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.
Protein design is the rational design of new protein molecules to design novel activity, behavior, or purpose, and to advance basic understanding of protein function. Proteins can be designed from scratch or by making calculated variants of a known protein structure and its sequence. Rational protein design approaches make protein-sequence predictions that will fold to specific structures. These predicted sequences can then be validated experimentally through methods such as peptide synthesis, site-directed mutagenesis, or artificial gene synthesis.
Lattice proteins are highly simplified models of protein-like heteropolymer chains on lattice conformational space which are used to investigate protein folding. Simplification in lattice proteins is twofold: each whole residue is modeled as a single "bead" or "point" of a finite set of types, and each residue is restricted to be placed on vertices of a lattice. To guarantee the connectivity of the protein chain, adjacent residues on the backbone must be placed on adjacent vertices of the lattice. Steric constraints are expressed by imposing that no more than one residue can be placed on the same lattice vertex.
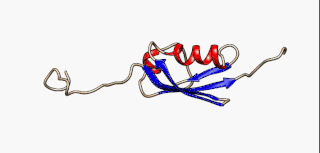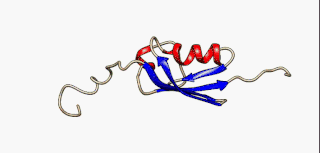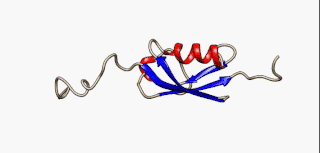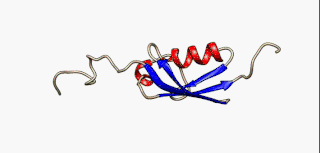
In molecular biology, an intrinsically disordered protein (IDP) is a protein that lacks a fixed or ordered three-dimensional structure, typically in the absence of its macromolecular interaction partners, such as other proteins or RNA. IDPs range from fully unstructured to partially structured and include random coil, molten globule-like aggregates, or flexible linkers in large multi-domain proteins. They are sometimes considered as a separate class of proteins along with globular, fibrous and membrane proteins.
Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.
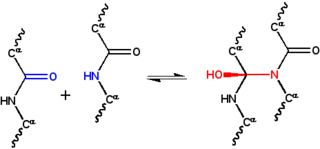
The cyclol hypothesis is the now discredited first structural model of a folded, globular protein, formulated in the 1930s. It was based on the cyclol reaction of peptide bonds proposed by physicist Frederick Frank in 1936, in which two peptide groups are chemically crosslinked. These crosslinks are covalent analogs of the non-covalent hydrogen bonds between peptide groups and have been observed in rare cases, such as the ergopeptides.
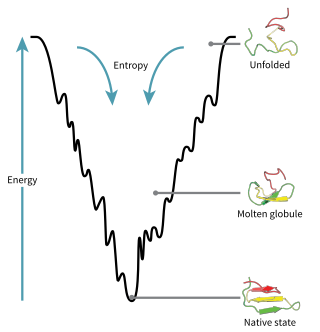
The folding funnel hypothesis is a specific version of the energy landscape theory of protein folding, which assumes that a protein's native state corresponds to its free energy minimum under the solution conditions usually encountered in cells. Although energy landscapes may be "rough", with many non-native local minima in which partially folded proteins can become trapped, the folding funnel hypothesis assumes that the native state is a deep free energy minimum with steep walls, corresponding to a single well-defined tertiary structure. The term was introduced by Ken A. Dill in a 1987 article discussing the stabilities of globular proteins.
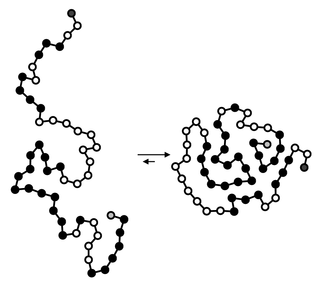
Hydrophobic collapse is a proposed process for the production of the 3-D conformation adopted by polypeptides and other molecules in polar solvents. The theory states that the nascent polypeptide forms initial secondary structure creating localized regions of predominantly hydrophobic residues. The polypeptide interacts with water, thus placing thermodynamic pressures on these regions which then aggregate or "collapse" into a tertiary conformation with a hydrophobic core. Incidentally, polar residues interact favourably with water, thus the solvent-facing surface of the peptide is usually composed of predominantly hydrophilic regions.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.
In molecular biology, a protein domain is a region of a protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains, and a domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length. The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred. Usually this common ancestry is inferred from structural alignment and mechanistic similarity, even if no sequence similarity is evident. Sequence homology can then be deduced even if not apparent. Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease and glycosyl hydrolases superfamilies based on the MEROPS and CAZy classification systems.

