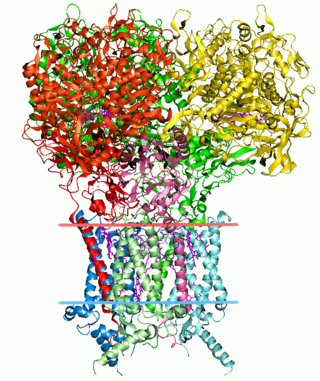
Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
A chemical database is a database specifically designed to store chemical information. This information is about chemical and crystal structures, spectra, reactions and syntheses, and thermophysical data.

Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
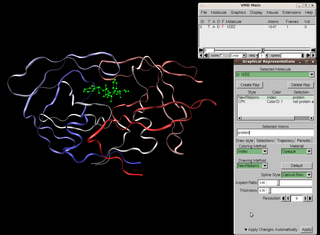
Visual Molecular Dynamics (VMD) is a molecular modelling and visualization computer program. VMD is developed mainly as a tool to view and analyze the results of molecular dynamics simulations. It also includes tools for working with volumetric data, sequence data, and arbitrary graphics objects. Molecular scenes can be exported to external rendering tools such as POV-Ray, RenderMan, Tachyon, Virtual Reality Modeling Language (VRML), and many others. Users can run their own Tcl and Python scripts within VMD as it includes embedded Tcl and Python interpreters. VMD runs on Unix, Apple Mac macOS, and Microsoft Windows. VMD is available to non-commercial users under a distribution-specific license which permits both use of the program and modification of its source code, at no charge.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
Protein–ligand docking is a molecular modelling technique. The goal of protein–ligand docking is to predict the position and orientation of a ligand when it is bound to a protein receptor or enzyme. Pharmaceutical research employs docking techniques for a variety of purposes, most notably in the virtual screening of large databases of available chemicals in order to select likely drug candidates. There has been rapid development in computational ability to determine protein structure with programs such as AlphaFold, and the demand for the corresponding protein-ligand docking predictions is driving implementation of software that can find accurate models. Once the protein folding can be predicted accurately along with how the ligands of various structures will bind to the protein, the ability for drug development to progress at a much faster rate becomes possible.
Modeller, often stylized as MODELLER, is a computer program used for homology modeling to produce models of protein tertiary structures and quaternary structures (rarer). It implements a method inspired by nuclear magnetic resonance spectroscopy of proteins, termed satisfaction of spatial restraints, by which a set of geometrical criteria are used to create a probability density function for the location of each atom in the protein. The method relies on an input sequence alignment between the target amino acid sequence to be modeled and a template protein which structure has been solved.
In molecular modelling, docking is a method which predicts the preferred orientation of one molecule to another when bound together in a stable complex. In the case of protein docking, the search space consists of all possible orientations of the protein with respect to the ligand. Flexible docking in addition considers all possible conformations of the protein paired with all possible conformations of the ligand.
In the fields of computational chemistry and molecular modelling, scoring functions are mathematical functions used to approximately predict the binding affinity between two molecules after they have been docked. Most commonly one of the molecules is a small organic compound such as a drug and the second is the drug's biological target such as a protein receptor. Scoring functions have also been developed to predict the strength of intermolecular interactions between two proteins or between protein and DNA.

Avogadro is a molecule editor and visualizer designed for cross-platform use in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It is extensible via a plugin architecture.
AutoDock is a molecular modeling simulation software. It is especially effective for protein-ligand docking. AutoDock 4 is available under the GNU General Public License. AutoDock is one of the most cited docking software applications in the research community. It is used by the FightAIDS@Home and OpenPandemics - COVID-19 projects run at World Community Grid, to search for antivirals against HIV/AIDS and COVID-19. In February 2007, a search of the ISI Citation Index showed more than 1,100 publications had been cited using the primary AutoDock method papers. As of 2009, this number surpassed 1,200.
Computational Resources for Drug Discovery (CRDD) is one of the important silico modules of Open Source for Drug Discovery (OSDD). The CRDD web portal provides computer resources related to drug discovery on a single platform. It provides computational resources for researchers in computer-aided drug design, a discussion forum, and resources to maintain a wiki related to drug discovery, predict inhibitors, and predict the ADME-Tox property of molecules. One of the major objectives of CRDD is to promote open source software in the field of chemoinformatics and pharmacoinformatics.
Lead Finder is a computational chemistry tool designed for modeling protein-ligand interactions. This application is useful for conducting molecular docking studies and quantitatively assessing ligand binding and biological activity. Lead Finder offers free access to individual users, especially those in non-commercial and academic settings.
CheShift-2 is an application created to compute 13Cα and 13Cβ protein chemical shifts and to validate protein structures. It is based on quantum mechanics computations of 13Cα and 13Cβchemical shift as a function of the torsional angles of the 20 amino acids.
LiSiCA is a ligand-based virtual screening software that searches for 2D and 3D similarities between a reference compound and a database of target compounds which should be represented in a Mol2 format. The similarities are expressed using the Tanimoto coefficients and the target compounds are ranked accordingly. LiSiCA is also available as LiSiCA PyMOL plugin both on Linux and Windows operating systems.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.
LeDock is a proprietary, flexible molecular docking software designed for the purpose of docking ligands with target proteins. It is available for Linux, macOS, and Windows.

rDock is an open-source molecular docking software that be used for docking small molecules against proteins and nucleic acids. It is primarily designed for high-throughput virtual screening and prediction of binding mode.
Namasivayam Gautham is a retired Professor Emeritus at the Centre of Advance Study in Crystallography and Biophysics, University of Madras. He is known for his work on DNA Crystallography, protein structure prediction and molecular docking.