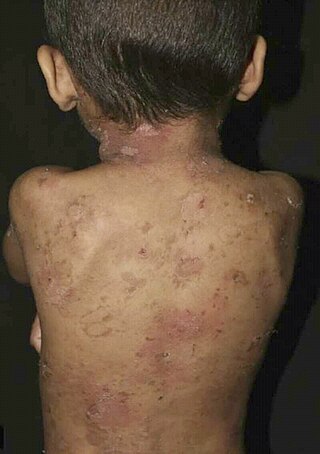
Epidermolytic ichthyosis (EI), is a severe form of dry scaly skin, that initially presents with redness, blisters, erosions, and peeling in a newborn baby. Hyperkeratosis typically develops several months later. Other symptoms include itch, painful fissures, strong body odor, and absence of sweat. Symptoms vary in severity and extent of skin involvement. The two main types are divided into one involving palms and soles and the other without.
Type II keratins constitutes the Type II intermediate filaments (IFs) of the intracytoplasmatic cytoskeleton, which is present in all mammalian epithelial cells. The type 2 cytokeratins consist of basic or neutral, high molecular weight proteins which in vivo are arranged in pairs of heterotypic Type I and Type II keratin chains, coexpressed during differentiation of simple and stratified epithelial tissues. It has been seen that Type II Keratins are developed before Type 1 keratins during human embryonic development.
Keratin 14 is a member of the type I keratin family of intermediate filament proteins. Keratin 14 was the first type I keratin sequence determined. Keratin 14 is also known as cytokeratin-14 (CK-14) or keratin-14 (KRT14). In humans it is encoded by the KRT14 gene.

Hemidesmosomes are very small stud-like structures found in keratinocytes of the epidermis of skin that attach to the extracellular matrix. They are similar in form to desmosomes when visualized by electron microscopy; however, desmosomes attach to adjacent cells. Hemidesmosomes are also comparable to focal adhesions, as they both attach cells to the extracellular matrix. Instead of desmogleins and desmocollins in the extracellular space, hemidesmosomes utilize integrins. Hemidesmosomes are found in epithelial cells connecting the basal epithelial cells to the lamina lucida, which is part of the basal lamina. Hemidesmosomes are also involved in signaling pathways, such as keratinocyte migration or carcinoma cell intrusion.
DEBRA is the name of an international medical research charity dedicated to securing effective drug treatments and ultimately cures for every type of epidermolysis bullosa, with national groups in over 40 countries including in the United Kingdom and the United States.
Jonathan "Jonny" Kennedy was a British man who had a rare inherited condition known as dystrophic epidermolysis bullosa. Kennedy ultimately died of skin cancer, a complication of EB.
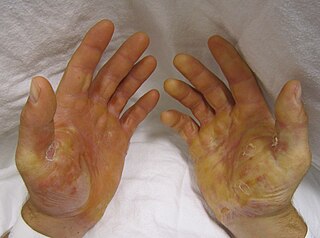
Epidermolysis bullosa simplex (EBS) is a disorder resulting from mutations in the genes encoding keratin 5 or keratin 14. It is one of the major forms of epidermolysis bullosa, a group of genetic conditions that cause the skin to be very fragile and to blister easily.

Kindler syndrome is a rare congenital disease of the skin caused by a mutation in the KIND1 gene.

Keratin 5, also known as KRT5, K5, or CK5, is a protein that is encoded in humans by the KRT5 gene. It dimerizes with keratin 14 and forms the intermediate filaments (IF) that make up the cytoskeleton of basal epithelial cells. This protein is involved in several diseases including epidermolysis bullosa simplex and breast and lung cancers.

Junctional epidermolysis bullosa (JEB) is an inherited disorder that is also known as red foot disease or hairless foal syndrome. JEB is the result of a genetic mutation that inhibits protein production that is essential for skin adhesion. Therefore, tissues, such as skin and mouth epithelia, are affected. Blisters form over the entire body causing pain and discomfort, and open sores leave newborn foals highly susceptible to secondary infection. The condition can be categorized into two types of mutations: JEB1 and JEB2. JEB1 is found in Belgian Draft horses, as well as other related Draft breeds. In contrast, JEB2 is found in American Saddlebred horses.

Epidermolysis bullosa dystrophica or dystrophic EB (DEB) is an inherited disease affecting the skin and other organs.

Genodermatosis is a hereditary skin disease with three inherited modes including single gene inheritance, multiple gene inheritance and chromosome inheritance. There are many different types of genodermatosis; the prevalence of genodermatosis ranges from 1 per 6000 people to 1 per 500,000 people. Genodermatosis has influence on the texture, color and structure of skin cuticle and connective tissue, specific lesion site and clinical manifestations on the body vary depending on the type. In the spite of the variety and complexity of genodermatosis, there are still some common methods that can help people diagnose. After diagnosis, different types of genodermatosis require different levels of therapy including interventions, nursing interventions and treatments. Among that, research of therapy for some new, complex and rare types are still in the developing stage. The impact of genodermatosis not only can be seen in body but also can be seen in all aspects of patients' life, including but not limited to psychological, family life, economic conditions and social activities. Accordingly, the patients need treatment, support and help in these areas.

Collagen alpha-1(VII) chain is a protein that in humans is encoded by the COL7A1 gene. It is composed of a triple helical, collagenous domain flanked by two non-collagenous domains, and functions as an anchoring fibril between the dermal-epidermal junction in the basement membrane. Mutations in COL7A1 cause all types of dystrophic epidermolysis bullosa, and the exact mutations vary based on the specific type or subtype. It has been shown that interactions between the NC-1 domain of collagen VII and several other proteins, including laminin-5 and collagen IV, contribute greatly to the overall stability of the basement membrane.
Anchoring fibrils extend from the basal lamina of epithelial cells and attach to the lamina reticularis by wrapping around the reticular fiber bundles. The basal lamina and lamina reticularis together make up the basement membrane. Anchoring fibrils are essential to the functional integrity of the dermoepidermal junction.
Junctional epidermolysis bullosa is a skin condition characterized by blister formation within the lamina lucida of the basement membrane zone.
Transient bullous dermolysis of the newborn (TBDN) is a skin condition that presents in newborns. It is characterized by blister formation secondary to even mild trauma.
A coma blister, or coma bullae, is a skin lesion or blister that typically arises due to pressure in an individual with impaired consciousness. They vary in size, ranging from 4 to 5 centimeters in diameter, and may appear hemorrhagic or blood filled. Coma blisters are usually found in the extremities and trunk. These types of blisters have been associated with the overdose of central nervous system (CNS) depressants especially barbiturates, but also tricyclic antidepressants, hypnotics, benzodiazepines, opiates, antipsychotics, and alcohol. However, studies have found that coma blisters are not caused by the toxicity of these drugs, but due to hypoxia and external pressure on the comatose individual's skin from being immobilized. Coma blisters have been frequently found on individuals who have overdosed on drugs, but have also been found on individuals with chronic kidney failure, hypercalcemia, diabetic ketoacidosis, and a variety of neurologic conditions. Coma blisters are more frequent in adults and less common among children as demonstrated by the few cases published in literature.
Scioderm, acquired by Amicus Therapeutics in 2015, was a rare disease company focused on developing a treatment for Epidermolysis Bullosa (EB), a rare genetic disease characterized by extremely fragile skin and recurrent blister formation. There are currently no approved therapies for EB. Scioderm was developing a topical treatment known as SD-101, or Zorblisa, aimed at triggering wound reduction and closure, and a reduction in body surface area coverage of blisters and lesions.
Beremagene geperpavec, sold under the brand name Vyjuvek, is a gene therapy for the treatment of wounds. Beremagene geperpavec is the first approved gene therapy to use herpes-simplex virus type 1 as a vector. Beremagene geperpavec is a genetically modified herpes-simplex virus used to deliver normal copies of the COL7A1 gene to the wounds.

Birch triterpenes, sold under the brand name Filsuvez, is an extract of birch bark used as a topical medication for the treatment of epidermolysis bullosa. The active ingredients are triterpenes extracted from the outer bark of silver birch and downy birch.


