Metalloprotein is a generic term for a protein that contains a metal ion cofactor. A large proportion of all proteins are part of this category. For instance, at least 1000 human proteins contain zinc-binding protein domains although there may be up to 3000 human zinc metalloproteins.
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.

A cofactor is a non-protein chemical compound or metallic ion that is required for an enzyme's role as a catalyst. Cofactors can be considered "helper molecules" that assist in biochemical transformations. The rates at which these happen are characterized in an area of study called enzyme kinetics. Cofactors typically differ from ligands in that they often derive their function by remaining bound.

Cytochromes P450 are a superfamily of enzymes containing heme as a cofactor that mostly, but not exclusively, function as monooxygenases. However, they are not omnipresent; for example, they have not been found in Escherichia coli. In mammals, these enzymes oxidize steroids, fatty acids, xenobiotics, and participate in many biosyntheses. By hydroxylation, CYP450 enzymes convert xenobiotics into hydrophilic derivatives, which are more readily excreted.
Cysteine dioxygenase (CDO) is a non-heme iron enzyme that catalyzes the conversion of L-cysteine to cysteine sulfinic acid. CDO plays an important role in cysteine catabolism, regulating intracellular levels of cysteine and responding changes in cysteine availability. As such, CDO is highly regulated and undergoes large changes in concentration and efficiency. It oxidizes cysteine to the corresponding sulfinic acid by activation of dioxygen, although the exact mechanism of the reaction is still unclear. In addition to being found in mammals, CDO also exists in some yeast and bacteria, although the exact function is still unknown. CDO has been implicated in various neurodegenerative diseases and cancers, which is likely related to cysteine toxicity.
Aromatic-ring-hydroxylating dioxygenases (ARHD) incorporate two atoms of dioxygen (O2) into their substrates in the dihydroxylation reaction. The product is (substituted) cis-1,2-dihydroxycyclohexadiene, which is subsequently converted to (substituted) benzene glycol by a cis-diol dehydrogenase.
An oxygenase is any enzyme that oxidizes a substrate by transferring the oxygen from molecular oxygen O2 (as in air) to it. The oxygenases form a class of oxidoreductases; their EC number is EC 1.13 or EC 1.14.

Catechol 1,2- dioxygenase is an enzyme that catalyzes the oxidative ring cleavage of catechol to form cis,cis-muconic acid:
In chemistry, a (redox) non-innocent ligand is a ligand in a metal complex where the oxidation state is not clear. Typically, complexes containing non-innocent ligands are redox active at mild potentials. The concept assumes that redox reactions in metal complexes are either metal or ligand localized, which is a simplification, albeit a useful one.
Formate dehydrogenases are a set of enzymes that catalyse the oxidation of formate to carbon dioxide, donating the electrons to a second substrate, such as NAD+ in formate:NAD+ oxidoreductase (EC 1.17.1.9) or to a cytochrome in formate:ferricytochrome-b1 oxidoreductase (EC 1.2.2.1). This family of enzymes has attracted attention as inspiration or guidance on methods for the carbon dioxide fixation, relevant to global warming.
Isopenicillin N synthase (IPNS) is a non-heme iron protein belonging to the 2-oxoglutarate (2OG)-dependent dioxygenases oxidoreductase family. This enzyme catalyzes the formation of isopenicillin N from δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine (LLD-ACV).
In enzymology, a phytanoyl-CoA dioxygenase (EC 1.14.11.18) is an enzyme that catalyzes the chemical reaction
In enzymology, a taurine dioxygenase (EC 1.14.11.17) is an enzyme that catalyzes the chemical reaction.
In enzymology, tryptophan 2,3-dioxygenase (EC 1.13.11.11) is a heme enzyme that catalyzes the oxidation of L-tryptophan (L-Trp) to N-formyl-L-kynurenine, as the first and rate-limiting step of the kynurenine pathway.
In enzymology, D-lysine 5,6-aminomutase is an enzyme that catalyzes the chemical reaction
A transition metal oxo complex is a coordination complex containing an oxo ligand. Formally O2-, an oxo ligand can be bound to one or more metal centers, i.e. it can exist as a terminal or (most commonly) as bridging ligands (Fig. 1). Oxo ligands stabilize high oxidation states of a metal. They are also found in several metalloproteins, for example in molybdenum cofactors and in many iron-containing enzymes. One of the earliest synthetic compounds to incorporate an oxo ligand is potassium ferrate (K2FeO4), which was likely prepared by Georg E. Stahl in 1702.
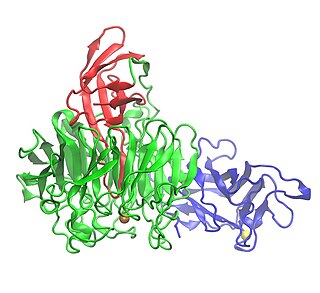
Galactose oxidase is an enzyme that catalyzes the oxidation of D-galactose in some species of fungi.
Alpha-ketoglutarate-dependent hydroxylases are a major class of non-heme iron proteins that catalyse a wide range of reactions. These reactions include hydroxylation reactions, demethylations, ring expansions, ring closures, and desaturations. Functionally, the αKG-dependent hydroxylases are comparable to cytochrome P450 enzymes. Both use O2 and reducing equivalents as cosubstrates and both generate water.
This article covers protein engineering of cytochrome (CYP) P450 enzymes. P450s are involved in a range of biochemical catabolic and anabolic process. Natural P450s can perform several different types of chemical reactions including hydroxylations, N,O,S-dealkylations, epoxidations, sulfoxidations, aryl-aryl couplings, ring contractions and expansions, oxidative cyclizations, alcohol/aldehyde oxidations, desaturations, nitrogen oxidations, decarboxylations, nitrations, as well as oxidative and reductive dehalogenations. Engineering efforts often strive for 1) improved stability 2) improved activity 3) improved substrate scope 4) enabled ability to catalyze unnatural reactions. P450 engineering is an emerging field in the areas of chemical biology and synthetic organic chemistry (chemoenzymatic).
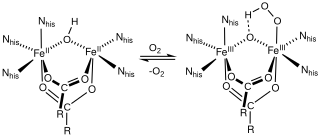
In biochemistry, non-heme iron proteins describe families of enzymes that utilize iron at the active site but lack heme cofactors. Iron-sulfur proteins, including those that are enzymes, are not included in this definition.







