
Hereditary haemochromatosis type 1 is a genetic disorder characterized by excessive intestinal absorption of dietary iron, resulting in a pathological increase in total body iron stores. Humans, like most animals, have no mechanism to regulate excess iron, simply losing a limited amount through various means like sweating or menstruating.
Chelation is a type of bonding of ions and their molecules to metal ions. It involves the formation or presence of two or more separate coordinate bonds between a polydentate ligand and a single central metal atom. These ligands are called chelants, chelators, chelating agents, or sequestering agents. They are usually organic compounds, but this is not a necessity.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.
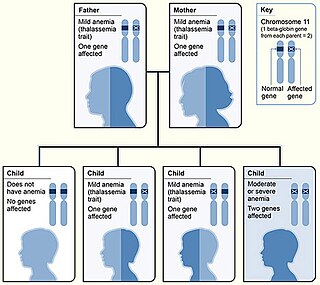
Thalassemias are inherited blood disorders that manifest as the production of reduced or zero quantities of hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe, including death. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Clinically, thalassemia is classed as Transfusion-Dependent Thalassemia (TDT) or non-Transfusion-Dependent Thalassemia (NTDT), since this determines the principal treatment options. TDT requires regular transfusions, typically every two to five weeks. TDTs include Beta-thalassemia major, non-deletional HbH disease, survived Hb Bart's disease, and severe HbE/beta-thalassemia. NTDT does not need regular transfusions but may require transfusion in case of an anemia crisis.

Iron overload is the abnormal and increased accumulation of total iron in the body, leading to organ damage. The primary mechanism of organ damage is oxidative stress, as elevated intracellular iron levels increase free radical formation via the Fenton reaction. Iron overload is often primary but may also be secondary to repeated blood transfusions. Iron deposition most commonly occurs in the liver, pancreas, skin, heart, and joints. People with iron overload classically present with the triad of liver cirrhosis, secondary diabetes mellitus, and bronze skin. However, due to earlier detection nowadays, symptoms are often limited to general chronic malaise, arthralgia, and hepatomegaly.

Chelation therapy is a medical procedure that involves the administration of chelating agents to remove heavy metals from the body. Chelation therapy has a long history of use in clinical toxicology and remains in use for some very specific medical treatments, although it is administered under very careful medical supervision due to various inherent risks, including the mobilization of mercury and other metals through the brain and other parts of the body by the use of weak chelating agents that unbind with metals before elimination, exacerbating existing damage. To avoid mobilization, some practitioners of chelation use strong chelators, such as selenium, taken at low doses over a long period of time.
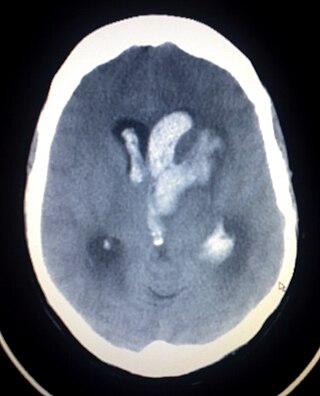
Intracerebral hemorrhage (ICH), also known as hemorrhagic stroke, is a sudden bleeding into the tissues of the brain, into its ventricles, or into both. An ICH is a type of bleeding within the skull and one kind of stroke. Symptoms can vary dramatically depending on the severity, acuity, and location (anatomically) but can include headache, one-sided weakness, numbness, tingling, or paralysis, speech problems, vision or hearing problems, memory loss, attention problems, coordination problems, balance problems, dizziness or lightheadedness or vertigo, nausea/vomiting, seizures, decreased level of consciousness or total loss of consciousness, neck stiffness, and fever.

Hepcidin is a protein that in humans is encoded by the HAMP gene. Hepcidin is a key regulator of the entry of iron into the circulation in mammals.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.

Deferasirox, sold under the brand name Exjade among others, is an oral iron chelator. Its main use is to reduce chronic iron overload in patients who are receiving long-term blood transfusions for conditions such as beta-thalassemia and other chronic anemias. It is the first oral medication approved in the United States for this purpose.

Platelet transfusion, is the process of infusing platelet concentrate into the body via vein, to prevent or treat the bleeding in people with either a low platelet count or poor platelet function. Often this occurs in people receiving cancer chemotherapy. Preventive transfusion is often done in those with platelet levels of less than 10 x 109/L. In those who are bleeding transfusion is usually carried out at less than 50 x 109/L. Blood group matching (ABO, RhD) is typically recommended before platelets are given. Unmatched platelets, however, are often used due to the unavailability of matched platelets. They are given by injection into a vein.
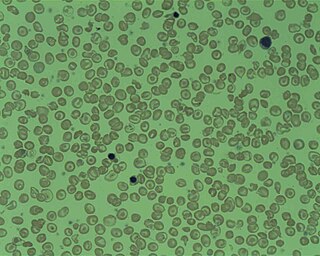
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

Deferiprone, sold under the brand name Ferriprox among others, is a medication that chelates iron and is used to treat iron overload in thalassaemia major. It was first approved and indicated for use in treating thalassaemia major in 1994 and had been licensed for use in the European Union for many years while awaiting approval in Canada and in the United States. On 14 October 2011, it was approved for use in the US under the FDA's accelerated approval program.
Transfusional hemosiderosis is the accumulation of iron in the body due to frequent blood transfusions. Iron accumulates in the liver and heart, but also endocrine organs. Frequent blood transfusions may be given to many patients, such as those with thalassemia, sickle cell disease, leukemia, aplastic anemia, or myelodysplastic syndrome, among others. It is diagnosed with a blood transferrin test and a liver biopsy. It is treated with venipuncture, erythrocytapheresis, and iron chelation therapy.
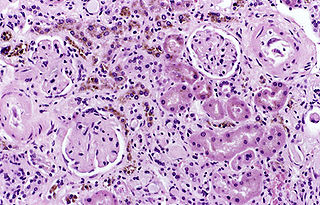
Hemosiderosis is a form of iron overload disorder resulting in the accumulation of hemosiderin.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CHA is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.
Congenital dyserythropoietic anemia (CDA) is a rare blood disorder, similar to the thalassemias. CDA is one of many types of anemia, characterized by ineffective erythropoiesis, and resulting from a decrease in the number of red blood cells (RBCs) in the body and a less than normal quantity of hemoglobin in the blood. CDA may be transmitted by both parents autosomal recessively or dominantly.

Susan Shurin is a senior adviser at the National Cancer Institute. From 2006–2014, she served as Deputy and Acting Director of the National Heart, Lung, and Blood Institute at the National Institutes of Health.
Treatment of the inherited blood disorder thalassemia depends upon the level of severity. For mild forms of the condition, advice and counseling are often all that are necessary. For more severe forms, treatment may consist in blood transfusion; chelation therapy to reverse iron overload, using drugs such as deferoxamine, deferiprone, or deferasirox; medication with the antioxidant indicaxanthin to prevent the breakdown of hemoglobin; or a bone marrow transplant using material from a compatible donor, or from the patient's mother. Removal of the spleen (splenectomy) could theoretically help to reduce the need for blood transfusions in people with thalassaemia major or intermedia but there is currently no reliable evidence from clinical trials about its effects. Population screening has had some success as a preventive measure.
Transfusion-dependent anemia is a form of anemia characterized by the need for continuous blood transfusion. It is a condition that results from various diseases, and is associated with decreased survival rates. Regular transfusion is required to reduce the symptoms of anemia by increasing functional red blood cells and hemoglobin count. Symptoms may vary based on the severity of the condition and the most common symptom is fatigue.
