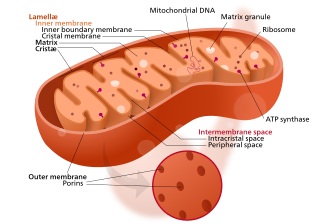
Mitochondrial membrane transport proteins, also known as mitochondrial carrier proteins, are proteins which exist in the membranes of mitochondria. They serve to transport molecules and other factors, such as ions, into or out of the organelles. Mitochondria contain both an inner and outer membrane, separated by the inter-membrane space, or inner boundary membrane. The outer membrane is porous, whereas the inner membrane restricts the movement of all molecules. The two membranes also vary in membrane potential and pH. These factors play a role in the function of mitochondrial membrane transport proteins. There are 53 discovered human mitochondrial membrane transporters, with many others that are known to still need discovered.

Mitofusin-2 is a protein that in humans is encoded by the MFN2 gene. Mitofusins are GTPases embedded in the outer membrane of the mitochondria. In mammals MFN1 and MFN2 are essential for mitochondrial fusion. In addition to the mitofusins, OPA1 regulates inner mitochondrial membrane fusion, and DRP1 is responsible for mitochondrial fission.
PTEN-induced kinase 1 (PINK1) is a mitochondrial serine/threonine-protein kinase encoded by the PINK1 gene.
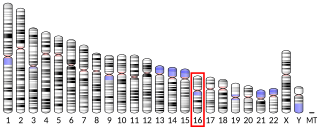
DnaJ homolog subfamily A member 3, mitochondrial, also known as Tumorous imaginal disc 1 (TID1), is a protein that in humans is encoded by the DNAJA3 gene on chromosome 16. This protein belongs to the DNAJ/Hsp40 protein family, which is known for binding and activating Hsp70 chaperone proteins to perform protein folding, degradation, and complex assembly. As a mitochondrial protein, it is involved in maintaining membrane potential and mitochondrial DNA (mtDNA) integrity, as well as cellular processes such as cell movement, growth, and death. Furthermore, it is associated with a broad range of diseases, including neurodegenerative diseases, inflammatory diseases, and cancers.

Dynamin-like 120 kDa protein, mitochondrial is a protein that in humans is encoded by the OPA1 gene. This protein regulates mitochondrial fusion and cristae structure in the inner mitochondrial membrane (IMM) and contributes to ATP synthesis and apoptosis, and small, round mitochondria. Mutations in this gene have been implicated in dominant optic atrophy (DOA), leading to loss in vision, hearing, muscle contraction, and related dysfunctions.

Mitochondrial fission 1 protein (FIS1) is a protein that in humans is encoded by the FIS1 gene on chromosome 7. This protein is a component of a mitochondrial complex, the ARCosome, that promotes mitochondrial fission. Its role in mitochondrial fission thus implicates it in the regulation of mitochondrial morphology, the cell cycle, and apoptosis. By extension, the protein is involved in associated diseases, including neurodegenerative diseases and cancers.
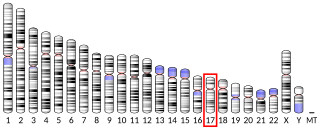
Mitochondrial Rho GTPase 1 (MIRO1) is an enzyme that in humans is encoded by the RHOT1 gene on chromosome 17. As a Miro protein isoform, the protein facilitates mitochondrial transport by attaching the mitochondria to the motor/adaptor complex. Through its key role in mitochondrial transport, RHOT1 is involved in mitochondrial homeostasis and apoptosis, as well as Parkinson's disease (PD) and cancer.

ATP-dependent metalloprotease YME1L1 is an enzyme that in humans is encoded by the YME1L1 gene. YME1L1 belongs to the AAA family of ATPases and mainly functions in the maintenance of mitochondrial morphology. Mutations in this gene would cause infantile-onset mitochondriopathy.
Interferon-induced guanylate-binding protein 2 is a protein that in humans is encoded by the GBP2 gene. GBP2 is a gene related to the superfamily of large GTPases which can be induced mainly by interferon gamma.
E3 ubiquitin-protein ligase MARCH5, also known as membrane-associated ring finger (C3HC4) 5, is an enzyme that, in humans, is encoded by the MARCH5 gene. It is localized in the mitochondrial outer membrane and has four transmembrane domains.
Mitophagy is the selective degradation of mitochondria by autophagy. It often occurs to defective mitochondria following damage or stress. The process of mitophagy was first described over a hundred years ago by Margaret Reed Lewis and Warren Harmon Lewis. Ashford and Porter used electron microscopy to observe mitochondrial fragments in liver lysosomes by 1962, and a 1977 report suggested that "mitochondria develop functional alterations which would activate autophagy." The term "mitophagy" was in use by 1998.
Mitochondrial biogenesis is the process by which cells increase mitochondrial numbers. It was first described by John Holloszy in the 1960s, when it was discovered that physical endurance training induced higher mitochondrial content levels, leading to greater glucose uptake by muscles. Mitochondrial biogenesis is activated by numerous different signals during times of cellular stress or in response to environmental stimuli, such as aerobic exercise.
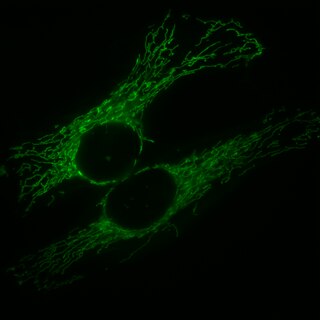
Mitochondrial fission is the process where mitochondria divide or segregate into two separate mitochondrial organelles. Mitochondrial fission is counteracted by the process of mitochondrial fusion, whereby two separate mitochondria can fuse together to form a large one. Mitochondrial fusion in turn can result in elongated mitochondrial networks. Both mitochondrial fission and fusion are balanced in the cell, and mutations interfering with either processes are associated with a variety of diseases. Mitochondria can divide by prokaryotic binary fission and since they require mitochondrial DNA for their function, fission is coordinated with DNA replication. Some of the proteins that are involved in mitochondrial fission have been identified and some of them are associated with mitochondrial diseases. Mitochondrial fission has significant implications in stress response and apoptosis.

Mitochondrial fission factor (Mff) is a protein that in humans is encoded by the MFF gene. Its primary role is in controlling the division of mitochondria. Mitochondrial morphology changes by continuous fission in order to create interconnected network of mitochondria. This activity is crucial for normal function of mitochondria. Mff is anchored to the mitochondrial outer membrane through the C-terminal transmembrane domain, extruding the bulk of the N-terminal portion containing two short amino acid repeats in the N-terminal half and a coiled-coil domain just upstream of the transmembrane domain into the cytosol. It has also been shown to regulate peroxisome morphology.
Mitochondria are dynamic organelles with the ability to fuse and divide (fission), forming constantly changing tubular networks in most eukaryotic cells. These mitochondrial dynamics, first observed over a hundred years ago are important for the health of the cell, and defects in dynamics lead to genetic disorders. Through fusion, mitochondria can overcome the dangerous consequences of genetic malfunction. The process of mitochondrial fusion involves a variety of proteins that assist the cell throughout the series of events that form this process.
Metalloendopeptidase OMA1, mitochondrial is an enzyme that in humans is encoded by the OMA1 gene. OMA1 is a Zn2+-dependent metalloendopeptidase in the inner membrane of mitochondria. The OMA1 acronym was derived from overlapping proteolytic activity with m-AAA protease 1.
Mitochondrial E3 ubiquitin protein ligase 1 (MUL1) is an enzyme that in humans is encoded by the MUL1 gene on chromosome 1. This enzyme localizes to the outer mitochondrial membrane, where it regulates mitochondrial morphology and apoptosis through multiple pathways, including the Akt, JNK, and NF-κB. Its proapoptotic function thus implicates it in cancer and Parkinson's disease.

The mycotoxin phomoxanthone A, or PXA for short, is a toxic natural product that affects the mitochondria. It is the most toxic and the best studied of the naturally occurring phomoxanthones. PXA has recently been shown to induce rapid, non-canonical mitochondrial fission by causing the mitochondrial matrix to fragment while the outer mitochondrial membrane can remain intact. This process was shown to be independent from the mitochondrial fission and fusion regulators DRP1 and OPA1.
Mitochondrial elongation factor 2 is a protein that in humans is encoded by the MIEF2 gene.

GFER syndrome is a rare mitochondrial disease. GFER was first reported in 2009 and since exome sequencing became more available, more cases were discovered. In all known cases, the disease progresses with conditions that include: congenital cataracts, loss of motor abilities, development delay, degeneration of organs, sometimes hearing loss, etc.




