In genetics, shotgun sequencing is a method used for sequencing random DNA strands. It is named by analogy with the rapidly expanding, quasi-random shot grouping of a shotgun.
Genomics is an interdisciplinary field of molecular biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.

A DNA sequencer is a scientific instrument used to automate the DNA sequencing process. Given a sample of DNA, a DNA sequencer is used to determine the order of the four bases: G (guanine), C (cytosine), A (adenine) and T (thymine). This is then reported as a text string, called a read. Some DNA sequencers can be also considered optical instruments as they analyze light signals originating from fluorochromes attached to nucleotides.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.

SOLiD (Sequencing by Oligonucleotide Ligation and Detection) is a next-generation DNA sequencing technology developed by Life Technologies and has been commercially available since 2006. This next generation technology generates 108 - 109 small sequence reads at one time. It uses 2 base encoding to decode the raw data generated by the sequencing platform into sequence data.

RNA-Seq is a technique that uses next-generation sequencing to reveal the presence and quantity of RNA molecules in a biological sample, providing a snapshot of gene expression in the sample, also known as transcriptome.
Cap analysis of gene expression (CAGE) is a gene expression technique used in molecular biology to produce a snapshot of the 5′ end of the messenger RNA population in a biological sample. The small fragments from the very beginnings of mRNAs are extracted, reverse-transcribed to cDNA, PCR amplified and sequenced. CAGE was first published by Hayashizaki, Carninci and co-workers in 2003. CAGE has been extensively used within the FANTOM research projects.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
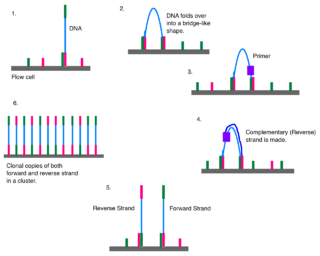
Illumina dye sequencing is a technique used to determine the series of base pairs in DNA, also known as DNA sequencing. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris. It was developed by Shankar Balasubramanian and David Klenerman of Cambridge University, who subsequently founded Solexa, a company later acquired by Illumina. This sequencing method is based on reversible dye-terminators that enable the identification of single nucleotides as they are washed over DNA strands. It can also be used for whole-genome and region sequencing, transcriptome analysis, metagenomics, small RNA discovery, methylation profiling, and genome-wide protein-nucleic acid interaction analysis.
In DNA sequencing, a read is an inferred sequence of base pairs corresponding to all or part of a single DNA fragment. A typical sequencing experiment involves fragmentation of the genome into millions of molecules, which are size-selected and ligated to adapters. The set of fragments is referred to as a sequencing library, which is sequenced to produce a set of reads.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.
G&T-seq is a novel form of single cell sequencing technique allowing one to simultaneously obtain both transcriptomic and genomic data from single cells, allowing for direct comparison of gene expression data to its corresponding genomic data in the same cell...

Duplex sequencing is a library preparation and analysis method for next-generation sequencing (NGS) platforms that employs random tagging of double-stranded DNA to detect mutations with higher accuracy and lower error rates.
Third-generation sequencing is a class of DNA sequencing methods which produce longer sequence reads, under active development since 2008.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.
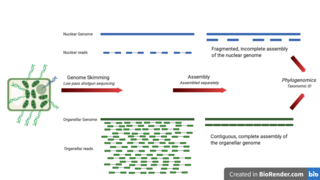
Genome skimming is a sequencing approach that uses low-pass, shallow sequencing of a genome, to generate fragments of DNA, known as genome skims. These genome skims contain information about the high-copy fraction of the genome. The high-copy fraction of the genome consists of the ribosomal DNA, plastid genome (plastome), mitochondrial genome (mitogenome), and nuclear repeats such as microsatellites and transposable elements. It employs high-throughput, next generation sequencing technology to generate these skims. Although these skims are merely 'the tip of the genomic iceberg', phylogenomic analysis of them can still provide insights on evolutionary history and biodiversity at a lower cost and larger scale than traditional methods. Due to the small amount of DNA required for genome skimming, its methodology can be applied in other fields other than genomics. Tasks like this include determining the traceability of products in the food industry, enforcing international regulations regarding biodiversity and biological resources, and forensics.
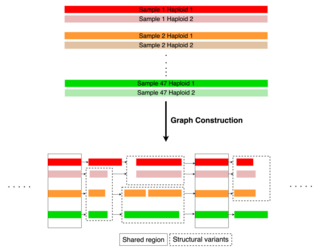
The Human Pangenome Reference is a collection of genomes from a diverse cohort of individuals compiled by the Human Pangenome Reference Consortium (HPRC). This first draft pangenome comprises 47 phased, diploid assemblies from a diverse cohort of individuals and was intended to capture the genetic diversity of the human population. The development of this pangenome seeks to address perceived shortcomings in the current human reference genome by offering a more comprehensive and inclusive resource for genomic research and analysis.
3' mRNA-seq is a quantitative, genome-wide transcriptomic technique based on the barcoding of the 3' untranslated region (UTR) of mRNA molecules. Unlike standard bulk RNA-seq, where short sequencing reads are generated along the entire length of mRNA transcripts, only the 3' end of polyadenylated RNAs are sequenced in 3' mRNA-seq. This approach results in a need for fewer reads to quantify the expression of a gene and reduces the sequencing depth required per sample while providing robust and reliable transcriptome-wide read-outs of gene expression levels comparable to full-length RNA-seq methods.

