WO2023069547A1 - 5-membered heteroaryl carboxamide compounds for treatment of hbv - Google Patents
5-membered heteroaryl carboxamide compounds for treatment of hbv Download PDFInfo
- Publication number
- WO2023069547A1 WO2023069547A1 PCT/US2022/047169 US2022047169W WO2023069547A1 WO 2023069547 A1 WO2023069547 A1 WO 2023069547A1 US 2022047169 W US2022047169 W US 2022047169W WO 2023069547 A1 WO2023069547 A1 WO 2023069547A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- mmol
- haloci
- chloro
- compound
- Prior art date
Links
- -1 5-membered heteroaryl carboxamide compounds Chemical class 0.000 title claims abstract description 170
- 238000011282 treatment Methods 0.000 title description 16
- 238000000034 method Methods 0.000 claims abstract description 93
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 22
- 208000015181 infectious disease Diseases 0.000 claims abstract description 19
- 208000002672 hepatitis B Diseases 0.000 claims abstract description 9
- 150000001875 compounds Chemical class 0.000 claims description 226
- 229910052739 hydrogen Inorganic materials 0.000 claims description 43
- 239000001257 hydrogen Substances 0.000 claims description 43
- 150000003839 salts Chemical class 0.000 claims description 41
- 150000002431 hydrogen Chemical group 0.000 claims description 28
- 125000000217 alkyl group Chemical group 0.000 claims description 23
- 229910052736 halogen Inorganic materials 0.000 claims description 21
- 150000002367 halogens Chemical group 0.000 claims description 21
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 20
- JKRDADVRIYVCCY-ZETCQYMHSA-N (2s)-2-hydroxyoctanoic acid Chemical compound CCCCCC[C@H](O)C(O)=O JKRDADVRIYVCCY-ZETCQYMHSA-N 0.000 claims description 19
- 125000001424 substituent group Chemical group 0.000 claims description 19
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 15
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 claims description 13
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 229910052799 carbon Inorganic materials 0.000 claims description 11
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 10
- 229910052717 sulfur Inorganic materials 0.000 claims description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 7
- 229910052740 iodine Inorganic materials 0.000 claims description 7
- 229910052794 bromium Inorganic materials 0.000 claims description 6
- 125000003342 alkenyl group Chemical group 0.000 claims description 5
- 125000000304 alkynyl group Chemical group 0.000 claims description 5
- 229910052801 chlorine Inorganic materials 0.000 claims description 4
- 229910052731 fluorine Inorganic materials 0.000 claims description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 4
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 3
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims 2
- 125000001475 halogen functional group Chemical group 0.000 claims 1
- 101710132601 Capsid protein Proteins 0.000 abstract description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 301
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 286
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 283
- 239000000243 solution Substances 0.000 description 156
- 239000000203 mixture Substances 0.000 description 139
- 239000011541 reaction mixture Substances 0.000 description 113
- 235000019439 ethyl acetate Nutrition 0.000 description 104
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 96
- 238000004809 thin layer chromatography Methods 0.000 description 87
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 83
- 239000007787 solid Substances 0.000 description 81
- 238000006243 chemical reaction Methods 0.000 description 78
- 239000012044 organic layer Substances 0.000 description 73
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 68
- 239000012043 crude product Substances 0.000 description 66
- 230000002829 reductive effect Effects 0.000 description 55
- ZMXDDKWLCZADIW-UHFFFAOYSA-N Vilsmeier-Haack reagent Natural products CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 52
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 51
- 238000005481 NMR spectroscopy Methods 0.000 description 47
- 238000004440 column chromatography Methods 0.000 description 47
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 40
- 239000000543 intermediate Substances 0.000 description 39
- 229910052938 sodium sulfate Inorganic materials 0.000 description 39
- 235000011152 sodium sulphate Nutrition 0.000 description 39
- 239000012267 brine Substances 0.000 description 38
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 38
- 238000002953 preparative HPLC Methods 0.000 description 34
- 238000010898 silica gel chromatography Methods 0.000 description 34
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 32
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 28
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 26
- 235000002639 sodium chloride Nutrition 0.000 description 24
- 239000003208 petroleum Substances 0.000 description 23
- 239000003826 tablet Substances 0.000 description 22
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 21
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 20
- 239000002775 capsule Substances 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 18
- 229910052786 argon Inorganic materials 0.000 description 16
- 239000012298 atmosphere Substances 0.000 description 15
- 230000015572 biosynthetic process Effects 0.000 description 15
- 239000000706 filtrate Substances 0.000 description 15
- 239000003112 inhibitor Substances 0.000 description 15
- 230000003612 virological effect Effects 0.000 description 15
- FYFWLJFQFXZGAH-UHFFFAOYSA-N ClC=1C=C(C=CC=1F)NC(=O)C1=C(N=CN1C)C1CC2CC(CC2C1)=O Chemical compound ClC=1C=C(C=CC=1F)NC(=O)C1=C(N=CN1C)C1CC2CC(CC2C1)=O FYFWLJFQFXZGAH-UHFFFAOYSA-N 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- 229920006395 saturated elastomer Polymers 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- 238000003786 synthesis reaction Methods 0.000 description 13
- 239000000463 material Substances 0.000 description 12
- 239000012299 nitrogen atmosphere Substances 0.000 description 12
- 238000004128 high performance liquid chromatography Methods 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 10
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 10
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 10
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 10
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- 235000019198 oils Nutrition 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- 108010050904 Interferons Proteins 0.000 description 9
- 102000014150 Interferons Human genes 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 229940079322 interferon Drugs 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 8
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 8
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 239000007821 HATU Substances 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 8
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- 125000003545 alkoxy group Chemical group 0.000 description 8
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 8
- 239000003937 drug carrier Substances 0.000 description 8
- 239000007924 injection Substances 0.000 description 8
- 238000002347 injection Methods 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 8
- 239000002253 acid Substances 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 239000002552 dosage form Substances 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 235000015320 potassium carbonate Nutrition 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- YWYRDBJNGFRAAB-UHFFFAOYSA-N BrC=1N=CN(C=1C(=O)NC1=CC(=C(C=C1)F)Cl)C Chemical compound BrC=1N=CN(C=1C(=O)NC1=CC(=C(C=C1)F)Cl)C YWYRDBJNGFRAAB-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 6
- 241000124008 Mammalia Species 0.000 description 6
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 5
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 5
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 5
- UZFRGJNXIPAGEX-UHFFFAOYSA-N BrC=1N(C(=C(N=1)Br)C(=O)NC1=CC(=C(C=C1)F)Cl)C Chemical compound BrC=1N(C(=C(N=1)Br)C(=O)NC1=CC(=C(C=C1)F)Cl)C UZFRGJNXIPAGEX-UHFFFAOYSA-N 0.000 description 5
- BTPZUGPPDOBDBQ-UHFFFAOYSA-N CN1C(C(NC(C=C2)=CC(Cl)=C2F)=O)=C(C(C2)CC(C3)C2CC3(C2=CC(Br)=NN2C)O)N=C1 Chemical compound CN1C(C(NC(C=C2)=CC(Cl)=C2F)=O)=C(C(C2)CC(C3)C2CC3(C2=CC(Br)=NN2C)O)N=C1 BTPZUGPPDOBDBQ-UHFFFAOYSA-N 0.000 description 5
- IBCXXLXICOMHBB-UHFFFAOYSA-N CN1C(C(NC(C=C2Cl)=CC=C2F)=O)=C(C(C2)CC(C3)C2CC3(C2=CC(O)=NN2C)O)N=C1 Chemical compound CN1C(C(NC(C=C2Cl)=CC=C2F)=O)=C(C(C2)CC(C3)C2CC3(C2=CC(O)=NN2C)O)N=C1 IBCXXLXICOMHBB-UHFFFAOYSA-N 0.000 description 5
- 108020004414 DNA Proteins 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 239000000443 aerosol Substances 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 230000000840 anti-viral effect Effects 0.000 description 5
- 210000000234 capsid Anatomy 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000011888 foil Substances 0.000 description 5
- 235000019253 formic acid Nutrition 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 125000005843 halogen group Chemical group 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 150000002430 hydrocarbons Chemical class 0.000 description 5
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 5
- 239000008101 lactose Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 239000006187 pill Substances 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 230000002441 reversible effect Effects 0.000 description 5
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 239000000829 suppository Substances 0.000 description 5
- 239000000454 talc Substances 0.000 description 5
- 235000012222 talc Nutrition 0.000 description 5
- 229910052623 talc Inorganic materials 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- UUFQTNFCRMXOAE-UHFFFAOYSA-N 1-methylmethylene Chemical compound C[CH] UUFQTNFCRMXOAE-UHFFFAOYSA-N 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 235000019270 ammonium chloride Nutrition 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 125000002837 carbocyclic group Chemical group 0.000 description 4
- 229920002301 cellulose acetate Polymers 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 229920001577 copolymer Polymers 0.000 description 4
- 239000013058 crude material Substances 0.000 description 4
- DQZKGSRJOUYVPL-UHFFFAOYSA-N cyclohexyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OC1CCCCC1 DQZKGSRJOUYVPL-UHFFFAOYSA-N 0.000 description 4
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 4
- 108020004707 nucleic acids Proteins 0.000 description 4
- 102000039446 nucleic acids Human genes 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 229920003023 plastic Polymers 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 238000003753 real-time PCR Methods 0.000 description 4
- 230000000707 stereoselective effect Effects 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000004808 supercritical fluid chromatography Methods 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 3
- KCDKSZWKVROYKI-UHFFFAOYSA-N (4-hydroxycyclohexyl) 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OC1CCC(O)CC1 KCDKSZWKVROYKI-UHFFFAOYSA-N 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 3
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 3
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- ORPPJRIAUMMMJR-UHFFFAOYSA-N 5-amino-N-(3-chloro-4-fluorophenyl)-3-[5-hydroxy-5-(methylsulfanylmethyl)-2,3,3a,4,6,6a-hexahydro-1H-pentalen-2-yl]-1-methylpyrazole-4-carboxamide Chemical compound NC1=C(C(=NN1C)C1CC2CC(CC2C1)(CSC)O)C(=O)NC1=CC(=C(C=C1)F)Cl ORPPJRIAUMMMJR-UHFFFAOYSA-N 0.000 description 3
- MZRUFMBFIKGOAL-UHFFFAOYSA-N 5-nitro-1h-pyrazole Chemical class [O-][N+](=O)C1=CC=NN1 MZRUFMBFIKGOAL-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- ZMASGVPZYXULFN-UHFFFAOYSA-N BrC=1C(=NN(C=1)CN1CCCC1)C(F)(F)F Chemical compound BrC=1C(=NN(C=1)CN1CCCC1)C(F)(F)F ZMASGVPZYXULFN-UHFFFAOYSA-N 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- 239000007818 Grignard reagent Substances 0.000 description 3
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 3
- 229930182821 L-proline Natural products 0.000 description 3
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 3
- PRYBSZCSRAGJIR-UHFFFAOYSA-N N-(3-chloro-4-fluorophenyl)-3-methylimidazole-4-carboxamide Chemical compound Cn1cncc1C(=O)Nc1ccc(F)c(Cl)c1 PRYBSZCSRAGJIR-UHFFFAOYSA-N 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 3
- 108091030071 RNAI Proteins 0.000 description 3
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 238000007112 amidation reaction Methods 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 239000003443 antiviral agent Substances 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 235000012216 bentonite Nutrition 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- QDGZDCVAUDNJFG-FXQIFTODSA-N entecavir (anhydrous) Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@H]1C[C@H](O)[C@@H](CO)C1=C QDGZDCVAUDNJFG-FXQIFTODSA-N 0.000 description 3
- 230000009368 gene silencing by RNA Effects 0.000 description 3
- 150000004795 grignard reagents Chemical class 0.000 description 3
- 125000001072 heteroaryl group Chemical group 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 229940043355 kinase inhibitor Drugs 0.000 description 3
- 239000000787 lecithin Substances 0.000 description 3
- 235000010445 lecithin Nutrition 0.000 description 3
- 229940067606 lecithin Drugs 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 150000007523 nucleic acids Chemical class 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 3
- 239000004033 plastic Substances 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 229960002429 proline Drugs 0.000 description 3
- 239000003380 propellant Substances 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 150000003254 radicals Chemical group 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 235000012239 silicon dioxide Nutrition 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- IQFYYKKMVGJFEH-CSMHCCOUSA-N telbivudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1O[C@@H](CO)[C@H](O)C1 IQFYYKKMVGJFEH-CSMHCCOUSA-N 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 3
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 3
- 235000019798 tripotassium phosphate Nutrition 0.000 description 3
- 229960005486 vaccine Drugs 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- ZNXOQNWQSFRXTL-UHFFFAOYSA-N 2-(3-nitropyrazol-1-yl)ethanol Chemical compound OCCN1C=CC([N+]([O-])=O)=N1 ZNXOQNWQSFRXTL-UHFFFAOYSA-N 0.000 description 2
- JPOAUGJNFIWTJB-UHFFFAOYSA-N 2-[(4-butoxyphenyl)methylsulfanyl]butanedioic acid Chemical compound CCCCOC1=CC=C(CSC(CC(O)=O)C(O)=O)C=C1 JPOAUGJNFIWTJB-UHFFFAOYSA-N 0.000 description 2
- CEBKHWWANWSNTI-UHFFFAOYSA-N 2-methylbut-3-yn-2-ol Chemical compound CC(C)(O)C#C CEBKHWWANWSNTI-UHFFFAOYSA-N 0.000 description 2
- YSEMCVGMNUUNRK-UHFFFAOYSA-N 3-chloro-4-fluoroaniline Chemical compound NC1=CC=C(F)C(Cl)=C1 YSEMCVGMNUUNRK-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- MJKVTPMWOKAVMS-UHFFFAOYSA-N 3-hydroxy-1-benzopyran-2-one Chemical compound C1=CC=C2OC(=O)C(O)=CC2=C1 MJKVTPMWOKAVMS-UHFFFAOYSA-N 0.000 description 2
- XGJYZAAIQYBIKF-UHFFFAOYSA-N 3-methylidenepyrrolidine Chemical compound C=C1CCNC1 XGJYZAAIQYBIKF-UHFFFAOYSA-N 0.000 description 2
- NAANUDVPTOXNLT-UHFFFAOYSA-N 4-bromo-1-(2-trimethylsilylethoxymethyl)pyrazole-3-carbaldehyde Chemical compound C[Si](C)(C)CCOCN1C=C(Br)C(C=O)=N1 NAANUDVPTOXNLT-UHFFFAOYSA-N 0.000 description 2
- HWYWVZDJOQXJHH-UHFFFAOYSA-N 4-bromo-5-(difluoromethyl)-1h-pyrazole Chemical compound FC(F)C=1NN=CC=1Br HWYWVZDJOQXJHH-UHFFFAOYSA-N 0.000 description 2
- JTHNMRUVJDWVMJ-UHFFFAOYSA-N 4-bromo-5-(trifluoromethyl)-1h-pyrazole Chemical compound FC(F)(F)C=1NN=CC=1Br JTHNMRUVJDWVMJ-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- ABAAGACTTVYVAX-UHFFFAOYSA-N CCOC(C(C)OC1=NN(C)C(C2(CC(CC(C3)C4=C(C(NC(C=C5Cl)=CC=C5F)=O)N(C)C=N4)C3C2)O)=C1)=O Chemical compound CCOC(C(C)OC1=NN(C)C(C2(CC(CC(C3)C4=C(C(NC(C=C5Cl)=CC=C5F)=O)N(C)C=N4)C3C2)O)=C1)=O ABAAGACTTVYVAX-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 description 2
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108020004638 Circular DNA Proteins 0.000 description 2
- RNDSTEJKEQDNQE-UHFFFAOYSA-N ClC=1C=C(C=CC=1F)NC(=O)C1=C(N=CN1C)C1CC2CC(CC2C1)(C1=CC(=NN1C)[N+](=O)[O-])O Chemical compound ClC=1C=C(C=CC=1F)NC(=O)C1=C(N=CN1C)C1CC2CC(CC2C1)(C1=CC(=NN1C)[N+](=O)[O-])O RNDSTEJKEQDNQE-UHFFFAOYSA-N 0.000 description 2
- QPNCIKBSCFEAKX-UHFFFAOYSA-N ClC=1C=C(C=CC=1F)NC(=O)C1=C(N=CN1C)C1CC2CC(CC2C1)(O)C#CC(=O)OC Chemical compound ClC=1C=C(C=CC=1F)NC(=O)C1=C(N=CN1C)C1CC2CC(CC2C1)(O)C#CC(=O)OC QPNCIKBSCFEAKX-UHFFFAOYSA-N 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 2
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 241000206672 Gelidium Species 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 241000700721 Hepatitis B virus Species 0.000 description 2
- 229930194542 Keto Natural products 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Substances BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- 108090001074 Nucleocapsid Proteins Proteins 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 229940044665 STING agonist Drugs 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 238000003477 Sonogashira cross-coupling reaction Methods 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 238000006069 Suzuki reaction reaction Methods 0.000 description 2
- 108010078233 Thymalfasin Proteins 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 108020000999 Viral RNA Proteins 0.000 description 2
- 229940118555 Viral entry inhibitor Drugs 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- YQNQNVDNTFHQSW-UHFFFAOYSA-N acetic acid [2-[[(5-nitro-2-thiazolyl)amino]-oxomethyl]phenyl] ester Chemical compound CC(=O)OC1=CC=CC=C1C(=O)NC1=NC=C([N+]([O-])=O)S1 YQNQNVDNTFHQSW-UHFFFAOYSA-N 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 230000008484 agonism Effects 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 150000001345 alkine derivatives Chemical class 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 230000009435 amidation Effects 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 230000008485 antagonism Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 229940121357 antivirals Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 238000012054 celltiter-glo Methods 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- GBBJCSTXCAQSSJ-XQXXSGGOSA-N clevudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1[C@H](F)[C@@H](O)[C@H](CO)O1 GBBJCSTXCAQSSJ-XQXXSGGOSA-N 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 229940097362 cyclodextrins Drugs 0.000 description 2
- 239000000134 cyclophilin inhibitor Substances 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 229940038472 dicalcium phosphate Drugs 0.000 description 2
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 210000001198 duodenum Anatomy 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 229960000980 entecavir Drugs 0.000 description 2
- 230000001973 epigenetic effect Effects 0.000 description 2
- IOLQWGVDEFWYNP-UHFFFAOYSA-N ethyl 2-bromo-2-methylpropanoate Chemical compound CCOC(=O)C(C)(C)Br IOLQWGVDEFWYNP-UHFFFAOYSA-N 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 2
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 2
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 2
- 210000003405 ileum Anatomy 0.000 description 2
- 229960001438 immunostimulant agent Drugs 0.000 description 2
- 239000003022 immunostimulating agent Substances 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 210000001630 jejunum Anatomy 0.000 description 2
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 2
- HQCYVSPJIOJEGA-UHFFFAOYSA-N methoxycoumarin Chemical compound C1=CC=C2OC(=O)C(OC)=CC2=C1 HQCYVSPJIOJEGA-UHFFFAOYSA-N 0.000 description 2
- FEXBHFZPLXHJAK-UHFFFAOYSA-N methyl 2,5-dibromo-3-methylimidazole-4-carboxylate Chemical compound COC(=O)C1=C(Br)N=C(Br)N1C FEXBHFZPLXHJAK-UHFFFAOYSA-N 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 2
- 239000002777 nucleoside Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 229960001860 salicylate Drugs 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 229940075439 smac mimetic Drugs 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000008247 solid mixture Substances 0.000 description 2
- 229940032147 starch Drugs 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- HHVIBTZHLRERCL-UHFFFAOYSA-N sulfonyldimethane Chemical compound CS(C)(=O)=O HHVIBTZHLRERCL-UHFFFAOYSA-N 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 229960005311 telbivudine Drugs 0.000 description 2
- SVUJNSGGPUCLQZ-FQQAACOVSA-N tenofovir alafenamide fumarate Chemical compound OC(=O)\C=C\C(O)=O.O([P@@](=O)(CO[C@H](C)CN1C2=NC=NC(N)=C2N=C1)N[C@@H](C)C(=O)OC(C)C)C1=CC=CC=C1.O([P@@](=O)(CO[C@H](C)CN1C2=NC=NC(N)=C2N=C1)N[C@@H](C)C(=O)OC(C)C)C1=CC=CC=C1 SVUJNSGGPUCLQZ-FQQAACOVSA-N 0.000 description 2
- 229960003560 tenofovir alafenamide fumarate Drugs 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 description 2
- 229960004231 thymalfasin Drugs 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- AQHMBDAHQGYLIU-XNFHFXFQSA-N (3s,6s,9s,12r,15s,18s,21s,24s,27r,30s,33s)-27-[2-(dimethylamino)ethylsulfanyl]-30-ethyl-33-[(e,1r,2r)-1-hydroxy-2-methylhex-4-enyl]-24-(2-hydroxy-2-methylpropyl)-1,4,7,10,12,15,19,25,28-nonamethyl-6,9,18-tris(2-methylpropyl)-3,21-di(propan-2-yl)-1,4,7,10, Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)(C)O)N(C)C(=O)[C@@H](SCCN(C)C)N(C)C1=O AQHMBDAHQGYLIU-XNFHFXFQSA-N 0.000 description 1
- QGKMIGUHVLGJBR-UHFFFAOYSA-M (4z)-1-(3-methylbutyl)-4-[[1-(3-methylbutyl)quinolin-1-ium-4-yl]methylidene]quinoline;iodide Chemical compound [I-].C12=CC=CC=C2N(CCC(C)C)C=CC1=CC1=CC=[N+](CCC(C)C)C2=CC=CC=C12 QGKMIGUHVLGJBR-UHFFFAOYSA-M 0.000 description 1
- QZLRSNPWLRXKTB-UHFFFAOYSA-N (6-formylpyridin-3-yl)boronic acid Chemical compound OB(O)C1=CC=C(C=O)N=C1 QZLRSNPWLRXKTB-UHFFFAOYSA-N 0.000 description 1
- 125000006645 (C3-C4) cycloalkyl group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- 108010030583 (melle-4)cyclosporin Proteins 0.000 description 1
- VXNQJPMCJMJOMN-UHFFFAOYSA-N 1,1-difluoroethane Chemical compound C[C](F)F VXNQJPMCJMJOMN-UHFFFAOYSA-N 0.000 description 1
- 125000004529 1,2,3-triazinyl group Chemical group N1=NN=C(C=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000004530 1,2,4-triazinyl group Chemical group N1=NC(=NC=C1)* 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000003363 1,3,5-triazinyl group Chemical group N1=C(N=CN=C1)* 0.000 description 1
- LYOKOJQBUZRTMX-UHFFFAOYSA-N 1,3-bis[[1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)propan-2-yl]oxy]-2,2-bis[[1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)propan-2-yl]oxymethyl]propane Chemical compound FC(F)(F)C(C(F)(F)F)(C(F)(F)F)OCC(COC(C(F)(F)F)(C(F)(F)F)C(F)(F)F)(COC(C(F)(F)F)(C(F)(F)F)C(F)(F)F)COC(C(F)(F)F)(C(F)(F)F)C(F)(F)F LYOKOJQBUZRTMX-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 1
- HSBMPUYCFQSKRP-UHFFFAOYSA-N 1-bromoimidazole Chemical compound BrN1C=CN=C1 HSBMPUYCFQSKRP-UHFFFAOYSA-N 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- KYWMCFOWDYFYLV-UHFFFAOYSA-N 1h-imidazole-2-carboxylic acid Chemical compound OC(=O)C1=NC=CN1 KYWMCFOWDYFYLV-UHFFFAOYSA-N 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- GCUOLJOTJRUDIZ-UHFFFAOYSA-N 2-(2-bromoethoxy)oxane Chemical compound BrCCOC1CCCCO1 GCUOLJOTJRUDIZ-UHFFFAOYSA-N 0.000 description 1
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- JBKINHFZTVLNEM-UHFFFAOYSA-N 2-bromoethoxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCCBr JBKINHFZTVLNEM-UHFFFAOYSA-N 0.000 description 1
- JNODDICFTDYODH-UHFFFAOYSA-N 2-hydroxytetrahydrofuran Chemical compound OC1CCCO1 JNODDICFTDYODH-UHFFFAOYSA-N 0.000 description 1
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- QWZHDKGQKYEBKK-UHFFFAOYSA-N 3-aminochromen-2-one Chemical compound C1=CC=C2OC(=O)C(N)=CC2=C1 QWZHDKGQKYEBKK-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- SITJXRWLFQGWCB-UHFFFAOYSA-N 3-iodo-1-methylpyrazole Chemical compound CN1C=CC(I)=N1 SITJXRWLFQGWCB-UHFFFAOYSA-N 0.000 description 1
- 125000004917 3-methyl-2-butyl group Chemical group CC(C(C)*)C 0.000 description 1
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 1
- PBEDVTDUVXFSMW-UHFFFAOYSA-N 3-methylimidazole-4-carboxylic acid Chemical compound CN1C=NC=C1C(O)=O PBEDVTDUVXFSMW-UHFFFAOYSA-N 0.000 description 1
- RTEVQEDKZPFGNP-UHFFFAOYSA-N 4-bromo-1-methylpiperidine Chemical compound CN1CCC(Br)CC1 RTEVQEDKZPFGNP-UHFFFAOYSA-N 0.000 description 1
- UWGFONONBAIDAF-UHFFFAOYSA-N 4-bromo-1h-pyrazole-5-carbaldehyde Chemical compound BrC=1C=NNC=1C=O UWGFONONBAIDAF-UHFFFAOYSA-N 0.000 description 1
- KKMFSVNFPUPGCA-UHFFFAOYSA-N 4-fluoro-3-(4-hydroxypiperidin-1-yl)sulfonyl-n-(3,4,5-trifluorophenyl)benzamide Chemical compound C1CC(O)CCN1S(=O)(=O)C1=CC(C(=O)NC=2C=C(F)C(F)=C(F)C=2)=CC=C1F KKMFSVNFPUPGCA-UHFFFAOYSA-N 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- BBLXLHYPDOMJMO-UHFFFAOYSA-N 5-(butan-2-ylsulfamoyl)-n-(3,4-difluorophenyl)-2-fluorobenzamide Chemical compound CCC(C)NS(=O)(=O)C1=CC=C(F)C(C(=O)NC=2C=C(F)C(F)=CC=2)=C1 BBLXLHYPDOMJMO-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 206010060933 Adverse event Diseases 0.000 description 1
- 239000012099 Alexa Fluor family Substances 0.000 description 1
- OLROWHGDTNFZBH-XEMWPYQTSA-N Alisporivir Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)N(CC)C(=O)[C@@H](C)N(C)C1=O OLROWHGDTNFZBH-XEMWPYQTSA-N 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 108700023418 Amidases Proteins 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- UIERETOOQGIECD-UHFFFAOYSA-N Angelic acid Natural products CC=C(C)C(O)=O UIERETOOQGIECD-UHFFFAOYSA-N 0.000 description 1
- 235000003276 Apios tuberosa Nutrition 0.000 description 1
- 101001053401 Arabidopsis thaliana Acid beta-fructofuranosidase 3, vacuolar Proteins 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000010744 Arachis villosulicarpa Nutrition 0.000 description 1
- PKWRMUKBEYJEIX-DXXQBUJASA-N Birinapant Chemical compound CN[C@@H](C)C(=O)N[C@@H](CC)C(=O)N1C[C@@H](O)C[C@H]1CC1=C(C2=C(C3=CC=C(F)C=C3N2)C[C@H]2N(C[C@@H](O)C2)C(=O)[C@H](CC)NC(=O)[C@H](C)NC)NC2=CC(F)=CC=C12 PKWRMUKBEYJEIX-DXXQBUJASA-N 0.000 description 1
- VPAYQCSPYASPNZ-UHFFFAOYSA-N BrC=1C(=NN(C=1)COCC[Si](C)(C)C)C(F)F Chemical compound BrC=1C(=NN(C=1)COCC[Si](C)(C)C)C(F)F VPAYQCSPYASPNZ-UHFFFAOYSA-N 0.000 description 1
- 229910014263 BrF3 Inorganic materials 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 102000001805 Bromodomains Human genes 0.000 description 1
- 108050009021 Bromodomains Proteins 0.000 description 1
- 238000007125 Buchwald synthesis reaction Methods 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- VFWAJOLBSRALMI-UHFFFAOYSA-N CC(C)(CNC1=NN(C)C(C2(CC(CC(C3)C4=C(C(NC(C=C5Cl)=CC=C5F)=O)N(C)C=N4)C3C2)O)=C1F)O Chemical compound CC(C)(CNC1=NN(C)C(C2(CC(CC(C3)C4=C(C(NC(C=C5Cl)=CC=C5F)=O)N(C)C=N4)C3C2)O)=C1F)O VFWAJOLBSRALMI-UHFFFAOYSA-N 0.000 description 1
- KJDLXOWKUWBTTQ-UHFFFAOYSA-N CN1C(=NC(=C1C(=O)O)Br)Br Chemical compound CN1C(=NC(=C1C(=O)O)Br)Br KJDLXOWKUWBTTQ-UHFFFAOYSA-N 0.000 description 1
- WUWJLWWTOLIXDH-UHFFFAOYSA-N CN1C(C(NC(C=C2Cl)=CC=C2F)=O)=C(C(C2)CC(C3)C2CC3(C2=CC(N)=NN2C)O)N=C1 Chemical compound CN1C(C(NC(C=C2Cl)=CC=C2F)=O)=C(C(C2)CC(C3)C2CC3(C2=CC(N)=NN2C)O)N=C1 WUWJLWWTOLIXDH-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- 229920008347 Cellulose acetate propionate Polymers 0.000 description 1
- DQEFEBPAPFSJLV-UHFFFAOYSA-N Cellulose propionate Chemical compound CCC(=O)OCC1OC(OC(=O)CC)C(OC(=O)CC)C(OC(=O)CC)C1OC1C(OC(=O)CC)C(OC(=O)CC)C(OC(=O)CC)C(COC(=O)CC)O1 DQEFEBPAPFSJLV-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 239000004859 Copal Substances 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 102000015792 Cyclin-Dependent Kinase 2 Human genes 0.000 description 1
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical class CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 102100036279 DNA (cytosine-5)-methyltransferase 1 Human genes 0.000 description 1
- FDTZUTSGGSRHQF-UHFFFAOYSA-N Desacetyl-nitazoxanide Chemical compound OC1=CC=CC=C1C(=O)NC1=NC=C([N+]([O-])=O)S1 FDTZUTSGGSRHQF-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 229920003143 Eudragit® FS 30 D Polymers 0.000 description 1
- 229920003139 Eudragit® L 100 Polymers 0.000 description 1
- 229920003138 Eudragit® L 30 D-55 Polymers 0.000 description 1
- 229920003141 Eudragit® S 100 Polymers 0.000 description 1
- LYJASUDJKPEXRN-UHFFFAOYSA-N FC=1C(=NN(C=1)C)I Chemical compound FC=1C(=NN(C=1)C)I LYJASUDJKPEXRN-UHFFFAOYSA-N 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 238000003747 Grignard reaction Methods 0.000 description 1
- 241000782205 Guibourtia conjugata Species 0.000 description 1
- 241000463109 Haloprofundus marisrubri Species 0.000 description 1
- 206010019799 Hepatitis viral Diseases 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 102100038970 Histone-lysine N-methyltransferase EZH2 Human genes 0.000 description 1
- 102100027704 Histone-lysine N-methyltransferase SETD7 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000931098 Homo sapiens DNA (cytosine-5)-methyltransferase 1 Proteins 0.000 description 1
- 101001028782 Homo sapiens Histone-lysine N-methyltransferase EZH1 Proteins 0.000 description 1
- 101000882127 Homo sapiens Histone-lysine N-methyltransferase EZH2 Proteins 0.000 description 1
- 101000650682 Homo sapiens Histone-lysine N-methyltransferase SETD7 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 229940083346 IAP antagonist Drugs 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102100040018 Interferon alpha-2 Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010079944 Interferon-alpha2b Proteins 0.000 description 1
- 102100021592 Interleukin-7 Human genes 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 108010028921 Lipopeptides Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 102000003959 Lymphotoxin-beta Human genes 0.000 description 1
- 108090000362 Lymphotoxin-beta Proteins 0.000 description 1
- 229940123628 Lysine (K)-specific demethylase 1A inhibitor Drugs 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- RJUFJBKOKNCXHH-UHFFFAOYSA-N Methyl propionate Chemical compound CCC(=O)OC RJUFJBKOKNCXHH-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 238000010934 O-alkylation reaction Methods 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 239000012661 PARP inhibitor Substances 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 108010090287 SCY-635 Proteins 0.000 description 1
- 101000615178 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) Serine/threonine-protein kinase SKY1 Proteins 0.000 description 1
- 102100031463 Serine/threonine-protein kinase PLK1 Human genes 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 102000011990 Sirtuin Human genes 0.000 description 1
- 108050002485 Sirtuin Proteins 0.000 description 1
- 239000004141 Sodium laurylsulphate Substances 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 102400000800 Thymosin alpha-1 Human genes 0.000 description 1
- 108010015780 Viral Core Proteins Proteins 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 229920002494 Zein Polymers 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- KDNSSKPZBDNJDF-UHFFFAOYSA-N [1-[(2-aminopurin-9-yl)methyl]cyclopropyl]oxymethylphosphonic acid Chemical compound C12=NC(N)=NC=C2N=CN1CC1(OCP(O)(O)=O)CC1 KDNSSKPZBDNJDF-UHFFFAOYSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- UTKBLLDLHPDWDU-ODZAUARKSA-N acetic acid;(z)-but-2-enedioic acid Chemical compound CC(O)=O.OC(=O)\C=C/C(O)=O UTKBLLDLHPDWDU-ODZAUARKSA-N 0.000 description 1
- IYKJEILNJZQJPU-UHFFFAOYSA-N acetic acid;butanedioic acid Chemical compound CC(O)=O.OC(=O)CCC(O)=O IYKJEILNJZQJPU-UHFFFAOYSA-N 0.000 description 1
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 1
- AEMQUICCWRPKDB-UHFFFAOYSA-N acetic acid;cyclohexane-1,2-dicarboxylic acid Chemical compound CC(O)=O.OC(=O)C1CCCCC1C(O)=O AEMQUICCWRPKDB-UHFFFAOYSA-N 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 229960003205 adefovir dipivoxil Drugs 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229940054685 alinia Drugs 0.000 description 1
- 108010058359 alisporivir Proteins 0.000 description 1
- 229950004789 alisporivir Drugs 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 102000005922 amidase Human genes 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 229940002637 baraclude Drugs 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 229950002782 besifovir Drugs 0.000 description 1
- 229940125388 beta agonist Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008236 biological pathway Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229950004237 birinapant Drugs 0.000 description 1
- 108010063132 birinapant Proteins 0.000 description 1
- 239000010836 blood and blood product Substances 0.000 description 1
- 229940125691 blood product Drugs 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 229940124765 capsid inhibitor Drugs 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 210000003855 cell nucleus Anatomy 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 229920006217 cellulose acetate butyrate Polymers 0.000 description 1
- 229920006218 cellulose propionate Polymers 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- 150000005827 chlorofluoro hydrocarbons Chemical class 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229960005338 clevudine Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- VKONPUDBRVKQLM-UHFFFAOYSA-N cyclohexane-1,4-diol Chemical compound OC1CCC(O)CC1 VKONPUDBRVKQLM-UHFFFAOYSA-N 0.000 description 1
- IBQKLMJCWQXCJK-UHFFFAOYSA-N cyclohexen-1-yl(dimethoxy)borane Chemical compound COB(OC)C1=CCCCC1 IBQKLMJCWQXCJK-UHFFFAOYSA-N 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 238000007256 debromination reaction Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 229940074057 epivir hbv Drugs 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- IRSJDVYTJUCXRV-UHFFFAOYSA-N ethyl 2-bromo-2,2-difluoroacetate Chemical compound CCOC(=O)C(F)(F)Br IRSJDVYTJUCXRV-UHFFFAOYSA-N 0.000 description 1
- ULNDTPIRBQGESN-UHFFFAOYSA-N ethyl 2-bromo-2-fluoroacetate Chemical compound CCOC(=O)C(F)Br ULNDTPIRBQGESN-UHFFFAOYSA-N 0.000 description 1
- ARFLASKVLJTEJD-UHFFFAOYSA-N ethyl 2-bromopropanoate Chemical compound CCOC(=O)C(C)Br ARFLASKVLJTEJD-UHFFFAOYSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- GDCRSXZBSIRSFR-UHFFFAOYSA-N ethyl prop-2-enoate;2-methylprop-2-enoic acid Chemical compound CC(=C)C(O)=O.CCOC(=O)C=C GDCRSXZBSIRSFR-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229940121360 farnesoid X receptor (fxr) agonists Drugs 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 102000034287 fluorescent proteins Human genes 0.000 description 1
- 108091006047 fluorescent proteins Proteins 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- NBVXSUQYWXRMNV-UHFFFAOYSA-N fluoromethane Chemical compound FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- UPBDXRPQPOWRKR-UHFFFAOYSA-N furan-2,5-dione;methoxyethene Chemical compound COC=C.O=C1OC(=O)C=C1 UPBDXRPQPOWRKR-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 229940097709 hepsera Drugs 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000002429 hydrazines Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- KJDJPXUIZYHXEZ-UHFFFAOYSA-N hydrogen sulfate;methylaminoazanium Chemical compound CN[NH3+].OS([O-])(=O)=O KJDJPXUIZYHXEZ-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000005113 hydroxyalkoxy group Chemical group 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229940090438 infergen Drugs 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 108010006088 interferon alfa-n1 Proteins 0.000 description 1
- 108010010648 interferon alfacon-1 Proteins 0.000 description 1
- 229940100994 interleukin-7 Drugs 0.000 description 1
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 229940065638 intron a Drugs 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 159000000014 iron salts Chemical class 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229960001627 lamivudine Drugs 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 238000007477 logistic regression Methods 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 210000004779 membrane envelope Anatomy 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- FVNJBPMQWSIGJK-HNNXBMFYSA-N methyl (4r)-4-(2-chloro-4-fluorophenyl)-2-(3,5-difluoropyridin-2-yl)-6-methyl-1,4-dihydropyrimidine-5-carboxylate Chemical compound C1([C@@H]2N=C(NC(C)=C2C(=O)OC)C=2C(=CC(F)=CN=2)F)=CC=C(F)C=C1Cl FVNJBPMQWSIGJK-HNNXBMFYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229940017219 methyl propionate Drugs 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- KXYJCOMKJRZAEN-UHFFFAOYSA-N n,n-dimethylformamide;ethanamine Chemical compound CCN.CN(C)C=O KXYJCOMKJRZAEN-UHFFFAOYSA-N 0.000 description 1
- OQIUTYABZMBBME-FMQUCBEESA-N n-[(e)-1-bromo-1-(2-methoxyphenyl)-3-oxo-3-piperidin-1-ylprop-1-en-2-yl]-4-nitrobenzamide Chemical compound COC1=CC=CC=C1\C(Br)=C(C(=O)N1CCCCC1)/NC(=O)C1=CC=C([N+]([O-])=O)C=C1 OQIUTYABZMBBME-FMQUCBEESA-N 0.000 description 1
- DJOVLOYCGXNVPI-UHFFFAOYSA-N n-[2-(4-methylpiperazin-1-yl)-5-[3-(morpholin-4-ylmethyl)phenyl]phenyl]-6-oxo-4-(trifluoromethyl)-1h-pyridine-3-carboxamide Chemical compound C1CN(C)CCN1C1=CC=C(C=2C=C(CN3CCOCC3)C=CC=2)C=C1NC(=O)C1=CNC(=O)C=C1C(F)(F)F DJOVLOYCGXNVPI-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000000025 natural resin Substances 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- RPJPZDVUUKWPGT-FOIHOXPVSA-N nim811 Chemical compound CC[C@H](C)[C@@H]1N(C)C(=O)CN(C)C(=O)[C@H](CC)NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC1=O RPJPZDVUUKWPGT-FOIHOXPVSA-N 0.000 description 1
- 229960002480 nitazoxanide Drugs 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 229960002969 oleic acid Drugs 0.000 description 1
- 235000021313 oleic acid Nutrition 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 238000012858 packaging process Methods 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229940002988 pegasys Drugs 0.000 description 1
- 108010092853 peginterferon alfa-2a Proteins 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 229940021222 peritoneal dialysis isotonic solution Drugs 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 108010056274 polo-like kinase 1 Proteins 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229920002744 polyvinyl acetate phthalate Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000012041 precatalyst Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- CAEWJEXPFKNBQL-UHFFFAOYSA-N prop-2-enyl carbonochloridate Chemical compound ClC(=O)OCC=C CAEWJEXPFKNBQL-UHFFFAOYSA-N 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical class CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- BXNMTOQRYBFHNZ-UHFFFAOYSA-N resiquimod Chemical compound C1=CC=CC2=C(N(C(COCC)=N3)CC(C)(C)O)C3=C(N)N=C21 BXNMTOQRYBFHNZ-UHFFFAOYSA-N 0.000 description 1
- 229950010550 resiquimod Drugs 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 229940080313 sodium starch Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical class [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- QAHVHSLSRLSVGS-UHFFFAOYSA-N sulfamoyl chloride Chemical compound NS(Cl)(=O)=O QAHVHSLSRLSVGS-UHFFFAOYSA-N 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- SGOIRFVFHAKUTI-ZCFIWIBFSA-N tenofovir (anhydrous) Chemical compound N1=CN=C2N(C[C@@H](C)OCP(O)(O)=O)C=NC2=C1N SGOIRFVFHAKUTI-ZCFIWIBFSA-N 0.000 description 1
- JJXOIFHXNBFRNV-UHFFFAOYSA-N tert-butyl (2-methylpropan-2-yl)oxycarbonyl carbonate Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C.CC(C)(C)OC(=O)OC(=O)OC(C)(C)C JJXOIFHXNBFRNV-UHFFFAOYSA-N 0.000 description 1
- CJVGFEARJOREHI-UHFFFAOYSA-N tert-butyl 3-(iodomethyl)azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC(CI)C1 CJVGFEARJOREHI-UHFFFAOYSA-N 0.000 description 1
- PXTONRTYYUAUJU-UHFFFAOYSA-N tert-butyl 3-methylidenepyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=C)C1 PXTONRTYYUAUJU-UHFFFAOYSA-N 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- UIERETOOQGIECD-ONEGZZNKSA-N tiglic acid Chemical compound C\C=C(/C)C(O)=O UIERETOOQGIECD-ONEGZZNKSA-N 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- SBUXRMKDJWEXRL-ZWKOTPCHSA-N trans-body Chemical compound O=C([C@@H]1N(C2=O)[C@H](C3=C(C4=CC=CC=C4N3)C1)CC)N2C1=CC=C(F)C=C1 SBUXRMKDJWEXRL-ZWKOTPCHSA-N 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 125000005591 trimellitate group Chemical group 0.000 description 1
- 229940063032 tyzeka Drugs 0.000 description 1
- KCFYEAOKVJSACF-UHFFFAOYSA-N umifenovir Chemical compound CN1C2=CC(Br)=C(O)C(CN(C)C)=C2C(C(=O)OCC)=C1CSC1=CC=CC=C1 KCFYEAOKVJSACF-UHFFFAOYSA-N 0.000 description 1
- 229960004626 umifenovir Drugs 0.000 description 1
- 235000019871 vegetable fat Nutrition 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 201000001862 viral hepatitis Diseases 0.000 description 1
- 210000000605 viral structure Anatomy 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 229940093612 zein Drugs 0.000 description 1
- 239000005019 zein Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 235000016804 zinc Nutrition 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 235000014692 zinc oxide Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Definitions
- HBV treatments can range from palliative to watchful waiting.
- Nucleotide analogs suppress virus production, treating the symptom, but leave the infection intact.
- Interferon a has severe side effects and less tolerability among patients and is successful as a finite treatment strategy in only a small minority of patients. There is a clear on-going need for more effective treatments for HBV infections.
- the disclosure provides a compound of Formula I: Formula I or a pharmaceutically acceptable salt thereof, where the variables are described in the detailed description.
- the disclosure provides a method of treating an HBV infection in a subject in need thereof, comprising: administering to the subject a therapeutically effective amount of compound of Formula I, or a pharmaceutically acceptable salt thereof.
- the disclosure provides a method of treating an HBV infection in a subject in need thereof, comprising: administering to the subject a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
- alkoxy refers to a straight or branched alkyl group attached to oxygen (i.e., alkyl-O-).
- alkoxy groups include, but are not limited to, alkoxy groups of 1-6 or 1-4 carbon atoms, referred to herein as Ci-ealkoxy and Ci-4alkoxy, respectively.
- alkoxy groups include, but are not limited to methoxy, ethoxy, and isopropoxy, etc....
- R 11 is independently selected for each occurrence from the group consisting of F, Cl, Br and I.
- X 1 is NR xl ; R xl is hydrogen or methyl; R 1 is hydrogen, OH or Ci-ealkoxy.
- compositions comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
- pharmaceutical compositions comprising compounds as disclosed herein formulated together with one or more pharmaceutically acceptable carriers.
- These formulations include those suitable for oral, rectal, topical, buccal, parenteral (e.g., subcutaneous, intramuscular, intradermal, or intravenous), rectal, vaginal, or aerosol administration, although the most suitable form of administration in any given case will depend on the degree and severity of the condition being treated and on the nature of the particular compound being used.
- disclosed compositions may be formulated as a unit dose, and/or may be formulated for oral or subcutaneous administration.
- Exemplary pharmaceutical compositions of this disclosure may be used in the form of a pharmaceutical preparation, for example, in solid, semisolid or liquid form, which contains one or more compounds of the disclosure, as an active ingredient, in admixture with an organic or inorganic carrier or excipient suitable for external, enteral or parenteral applications.
- the active ingredient may be compounded, for example, with the usual nontoxic, pharmaceutically acceptable carriers for tablets, pellets, capsules, suppositories, solutions, emulsions, suspensions, and any other form suitable for use.
- the active object compound is included in the pharmaceutical composition in an amount sufficient to produce the desired effect upon the process or condition of the disease.
- a disclosed compound may be administered as part of a combination therapy in conjunction with one or more antivirals.
- Example antivirals include nucleoside analogs, interferon a, and other assembly effectors, for instance heteroaryldihydropyrimidines (HAPs) such as methyl 4-(2-chloro-4-fluorophenyl)-6-methyl- 2-(pyridin-2-yl)-l,4-dihydropyrimidine-5-carboxylate (HAP-1).
- HAPs heteroaryldihydropyrimidines
- a method of treating a patient suffering from hepatitis B infection comprising administering to the patient a first amount of a disclosed compound and a second amount of an antiviral, or other anti HBV agent, for example a second amount of a second compound selected from the group consisting of: an HBV capsid assembly promoter (for example,
- HBc directed transbodies such as those described in Wang Y, et al, Transbody against hepatitis B virus core protein inhibits hepatitis B virus replication in vitro, Int.
- OICR-9429 OICR-9429
- PARP inhibitors APE inhibitors, DNMT inhibitors, LSD1 inhibitors, JMJD HDM inhibitors, and Bromodomain antagonists
- kinase inhibitors such as TKB1 antagonists, PLK1 inhibitors, SRPK inhibitors, CDK2 inhibitors, ATM & ATR kinase inhibitors
- STING Agonists Ribavirin; N-acetyl cysteine ; NOV-205 (BAM205); Nitazoxanide (Alinia), Tizoxanide; SB 9200 Small Molecule Nucleic Acid Hybrid (SMNH); DV-601; Arbidol; FXR agonists (such as GW 4064 and Fexaramin); antibodies, therapeutic proteins, gene therapy, and biologies directed against viral components or interacting host proteins.
- kinase inhibitors such as TKB1 antagonists, PLK1 inhibitors, SRPK inhibitors, CDK2 inhibitors,
- Coumpounds of general structure VII-3 and VII-5 can be synthesized according to the method shown in Scheme VII. 1-9 can be reached with an appropriately substituted 3-nitro pyrazole to form VII- 1 which can be reduced to yield VII-2. Amidation of VII-2 provide VII- 3. Halogentation of VII-2 yields VII-4, which can likewise be amidated obtain VII-5.
- Method B X-select CSH 18 (3 x 50 mm x 2.5pm); Mobile phase: A; 0.025% formic acid in H2O; B; CH3CN; Injection voloume:2 uL; Flow rate: 1.2 mL/min, column temperature: 50 °C; Gradient program: 0% B to 98% B in 2 min, hold until 3 min, at 3.2 min B cone, is 0 % until 4 min.
- Wells of 96-well clear bottom TC plate (Coming, cat#: 3904,) were seeded at 50,000 cells/well in 180 pL of treatment medium, and 20 pL of either 10% DMSO (Sigma, cat#: D4540) as controls or a 10X solution of test compounds in 10% DMSO in treatment media was added for a final compound concentration starting at 10 pM, and plates were incubated in 5% CO2 incubator at 37°C for 5 days.
- 10% DMSO Sigma, cat#: D4540
- 10X solution of test compounds in 10% DMSO in treatment media was added for a final compound concentration starting at 10 pM, and plates were incubated in 5% CO2 incubator at 37°C for 5 days.
- a crystal with size of 0.08 x 0.10 x 0.20mm of compound AIA-227-2 was obtained from EtOH after 20 days of volatilization and was used for X-ray diffraction data collection.
- the crystal belongs to monoclinic crystal system, with a space group P2i/c.
- the structure was solved by direct methods and all of the non-H atoms were refined against F by full-matrix least-squares methods using the SHELXTL program. All H atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms. Multi-scans absorption correction method was used, and the maximum and minimum transmission parameters were 0.7531 and 0.6017, respectively. The final R, wRi, GOF are 0.0457, 0.1293 and 1.024, respectively.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present disclosure provides, in part, 5-membered heteroaryl carboxamide compounds, and pharmaceutical compositions thereof, useful for disruption of HBV core protein assembly, and methods of treating Hepatitis B (HBV) infection.
Description
5-MEMBERED HETEROARYL CARBOXAMIDE COMPOUNDS FOR TREATMENT OF HBV
CROSS-REFERENCE TO RELATED APPLICATION
This application claims the benefit of U.S. Provisional Application No. 62/257,776, filed October 20, 2021, the contents of which are hereby incorporated by reference.
BACKGROUND
Hepatitis B (HBV) causes viral hepatitis that can further lead to chronic liver disease and increase the risk of liver cirrhosis and liver cancer (hepatocellular carcinoma). Worldwide, about 2 billion people have been infected with HBV, around 360 million people are chronically infected, and every year HBV infection causes more than one half million deaths. HBV can be spread by body fluids: from mother to child, by sex, and via blood products. Children bom to HBV-positive mothers may also be infected, unless vaccinated at birth.
The hepatitis virus particle is composed of a lipid envelope studded with surface protein (HBsAg) that surrounds the viral core. The core is composed of a protein shell, or capsid, built of 120 core protein (Cp) dimers, which in turn contains the relaxed circular DNA (rcDNA) viral genome as well as viral and host proteins. In an infected cell, the genome is found as a covalently closed circular DNA (cccDNA) in the host cell nucleus. The cccDNA is the template for viral RNAs and thus viral proteins. In the cytoplasm, Cp assembles around a complex of full-length viral RNA (the so-called pregenomic RNA or pgRNA and viral polymerase (P). After assembly, P reverse transcribes the pgRNA to rcDNA within the confines of the capsid to generate the DNA-filled viral core.
At present, chronic HBV is primarily treated with nucleos(t)ide analogs (e.g., entecavir) that suppress the vims while the patient remains on treatment, but do not eliminate the infection, even after many years of treatment. Once a patient starts taking nucleos(t)ide analogs, most must continue taking them or risk the possibility of a life-threatening immune response due to viral rebound. Further, nucleotide therapy may lead to the emergence of antiviral drug resistance.
The only FDA approved alternative to nucleos(t)ide analogs is treatment with interferon a or pegylated interferon a. Unfortunately, the adverse event incidence and profile of interferon a can result in poor tolerability, and many patients are unable to complete therapy. Moreover, only a small percentage of patients are considered appropriate for
interferon therapy, as only a small subset of patients is likely to have a sustained clinical response to a course of interferon therapy. As a result, interferon-based therapies are used in only a small percentage of all diagnosed patients who elect treatment.
Thus, current HBV treatments can range from palliative to watchful waiting. Nucleotide analogs suppress virus production, treating the symptom, but leave the infection intact. Interferon a has severe side effects and less tolerability among patients and is successful as a finite treatment strategy in only a small minority of patients. There is a clear on-going need for more effective treatments for HBV infections.
SUMMARY
The present disclosure provides, in part, 5-membed heteroaryl carboxamide compounds and pharmaceutical compositions thereof, useful for disruption of HB V core protein assembly, and methods of treating HBV infections.
In one aspect, the disclosure provides a compound of Formula I:
 Formula I or a pharmaceutically acceptable salt thereof, where the variables are described in the detailed description.
Formula I or a pharmaceutically acceptable salt thereof, where the variables are described in the detailed description.
In another aspect, the disclosure provides pharmaceutical compositions comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
In another aspect, the disclosure provides a method of treating an HBV infection in a subject in need thereof, comprising: administering to the subject a therapeutically effective amount of compound of Formula I, or a pharmaceutically acceptable salt thereof.
In another aspect, the disclosure provides a method of treating an HBV infection in a subject in need thereof, comprising: administering to the subject a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
BRIEF DESCRIPTION OF DRAWINGS
FIGURE 1 shows the ORTEP plot for compound CP-AIA-227-2.
FIGURE 2 shows the relative stereochemistry scheme of compound CP- AIA- 227-2.
DETAILED DESCRIPTION
The features and other details of the disclosure will now be more particularly described. Before further description of the present disclosure, certain terms employed in the specification, examples and appended claims are collected here. These definitions should be read in light of the remainder of the disclosure and as understood by a person of skill in the art. Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by a person of ordinary skill in the art.
I. Definitions
The term “alkenyl” as used herein refers to an unsaturated straight or branched hydrocarbon having at least one carbon-carbon double bond. Exemplary alkenyl groups include, but are not limited to, a straight or branched group of 2-6 carbon atoms, referred to herein as C2-ealkenyl. Exemplary alkenyl groups include, but are not limited to, vinyl, allyl, butenyl, and pentenyl, etc.
The term “alkoxy” as used herein refers to a straight or branched alkyl group attached to oxygen (i.e., alkyl-O-). Exemplary alkoxy groups include, but are not limited to, alkoxy groups of 1-6 or 1-4 carbon atoms, referred to herein as Ci-ealkoxy and Ci-4alkoxy, respectively. Exemplary alkoxy groups include, but are not limited to methoxy, ethoxy, and isopropoxy, etc....
The term “alkoxyalkyl” as used herein refers to an alkyl group substituted with an alkoxy group. Examples include, but are not limited to, CH3CH2OCH2-, CH3OCH2CH2- and CH3OCH2-, etc....
The term “alkyl” as used herein refers to a saturated straight or branched hydrocarbon. Exemplary alkyl groups include, but are not limited to, straight or branched hydrocarbons of
1-6 or 1-4 carbon atoms, referred to herein as C1-6 alkyl and CM alkyl, respectively. Exemplary alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, 2- methyl-1 -butyl, 3-methyl-2-butyl, 2-methyl-l-pentyl, 3-methyl-l-pentyl, 4-methyl-l -pentyl,
2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl, 2,2-dimethyl-l -butyl, 3,3- dimethyl-1 -butyl, 2-ethyl-l -butyl, n-butyl, isobutyl, t-butyl, n-pentyl, isopentyl, neopentyl, and n-hexyl, etc. The term “alkylene” as used herein refers to a biradical alkyl group.
The term “alkynyl” as used herein refers to an unsaturated straight or branched hydrocarbon having at least one carbon-carbon triple bond. Exemplary alkynyl groups include, but are not limited to, straight or branched groups of 2-6 carbon atoms, referred to herein as C2-6alkynyl. Exemplary alkynyl groups include, but are not limited to, ethynyl, propynyl, butynyl, pentynyl, hexynyl, and methylpropynyl, etc....
The term “carbonyl” as used herein refers to the biradical -C(O)-.
The term “cyano” as used herein refers to the radical -CN.
The terms “halo” or “halogen” as used herein refer to F, Cl, Br or I.
The term “haloalkyl” as used herein refers to an alkyl group substituted with one or more halogen atoms. For example, haloCi-ealkyl refers to a straight or branched alkyl group of 1-6 carbon atoms substituted with one or more halogen atoms. Examples include, but are not limited to, CH2F-, CHCh-, -CHF2, CF3-, CF3CH2-, CH3CF2, CF3CC12- and CF3CF2-.
The term “haloalkoxy” as used herein refers to an alkoxy group substituted with one or more halogen atoms. Examples include, but are not limited to, CC13O-, CF3O-, CHF2O- CF3CH2O-, and CF3CF2O-.
The term “heteroaryl” as used herein refers to a 5-6 membered monocyclic aromatic ring system containing one to four independently selected heteroatoms, such as nitrogen, oxygen and sulfur. Where possible, the heteroaryl ring may be linked to the adjacent radical though carbon or nitrogen. Examples of 5-6 membered monocyclic heteroaryl groups include, but are not limited to, furanyl, thiophenyl (also referred to as thienyl), pyrrolyl, thiazolyl, oxazolyl, isothiazolyl, isoxazolyl, imidazolyl, pyrazolyl, lH-l,2,3-triazolyl, 2H-
1.2.3-triazolyl, 1,2,4-triazolyl, pyridinyl (also referred to as pyridyl), pyridazinyl, pyrimidinyl, pyrazinyl, 1,3,5-triazinyl, 1,2,4-triazinyl, 1,2,3-triazinyl, 1,2,4-oxadiazolyl,
1.3.4-oxadiazolyl, 1,2,5-oxadiazolyl, 1,2,4-thiadiazolyl, 1,3,4-thiadiazolyl, 1,2,5-thiadiazolyl and tetrazolyl.
The terms “hydroxy” and “hydroxyl” as used herein refers to the radical -OH.
The term “hydroxyalkyl” as used herein refers to an alkyl group substituted with one or more hydroxy groups. Examples include, but are not limited to, HOCH2-, HOCH2CH2-, CH3CH(OH)CH2- and HOCH2CH(OH)CH2-.
The term “hydroxyalkoxy” as used herein refers to an alkoxy group substituted with one or more hydroxy groups. Examples include but are not limited to HOCH2O-, HOCH2CH2O-, CH3CH(OH)CH2O- and HOCH2CH(OH)CH2O-.
The term “RaRbNCi-6 alkyl-,” as used herein refers to an alkyl group substituted with a RaRbN- group, as defined herein. Examples include but are not limited to NH2CH2-, NH(CH3)CH2-, N(CH3)2CH2CH2- and CH3CH(NH2)CH2-.
The term “RaRbNCi-6alkoxy,” as used herein refers to an alkoxy group substituted with a RaRbN- groups, as defined herein. Examples include but are not limited to NH2CH2-, NH(CH3)CH2O-, N(CH3)2CH2CH2O-, and CH3CH(NH2)CH2O-.
The term “oxo” as used herein refers to the radical =0.
As used herein, when a bicyclic ring is shown with a floating point of attachment and/or floating substituents, for example
 signifies that the bicyclic ring can be attached via a carbon atom on either ring, and that the substituents (e.g., the R33 group(s)) can be independently attached to either or both rings.
signifies that the bicyclic ring can be attached via a carbon atom on either ring, and that the substituents (e.g., the R33 group(s)) can be independently attached to either or both rings.
The terms “Individual,” “patient,” or “subject” are used interchangeably and include any animal, including mammals, preferably mice, rats, other rodents, rabbits, dogs, cats, swine, cattle, sheep, horses, or primates, and most preferably humans. The compounds or pharmaceutical compositions of the disclosure can be administered to a mammal, such as a human, but can also be administered to other mammals such as an animal in need of veterinary treatment, e.g., domestic animals (e.g., dogs, cats, and the like), farm animals (e.g., cows, sheep, pigs, horses, and the like) and laboratory animals (e.g., rats, mice, guinea pigs, dogs, primates, and the like). The mammal treated in the methods of the disclosure is desirably a mammal in which treatment of HBV infection is desired.
The term “modulation” includes antagonism (e.g., inhibition), agonism, partial antagonism and/or partial agonism.
The term “Pharmaceutically acceptable” include molecular entities and compositions that do not produce an adverse, allergic or other untoward reaction when administered to an animal, or a human, as appropriate. For human administration, preparations should meet sterility, pyrogenicity, and general safety and purity standards as required by FDA Office of Biologies standards.
The term “pharmaceutically acceptable carrier” or “pharmaceutically acceptable excipient” as used herein refers to any and all solvents, dispersion media, coatings, isotonic and absorption delaying agents, fillers, and the like, that are compatible with pharmaceutical administration. The use of such media and agents for pharmaceutically active substances is
well known in the art. The compositions may also contain other active compounds providing supplemental, additional, or enhanced therapeutic functions.
The term “pharmaceutical composition” as used herein refers to a composition comprising at least one compound as disclosed herein formulated together with one or more pharmaceutically acceptable excipients.
The term "pharmaceutically acceptable salt(s)" as used herein refers to salts of acidic or basic groups that may be present in compounds used in the compositions. Compounds included in the present compositions that are basic in nature are capable of forming a wide variety of salts with various inorganic and organic acids. The acids that may be used to prepare pharmaceutically acceptable acid addition salts of such basic compounds are those that form non-toxic acid addition salts, i.e., salts containing pharmacologically acceptable anions, including, but not limited to, malate, oxalate, chloride, bromide, iodide, nitrate, sulfate, bisulfate, phosphate, acid phosphate, isonicotinate, acetate, lactate, salicylate, citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucaronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate (i.e., I , I '-methylene-/v.s-(2 -hydroxy-3 -naphthoate)) salts. Compounds included in the present compositions that are acidic in nature are capable of forming base salts with various pharmacologically acceptable cations. Examples of such salts include alkali metal or alkaline earth metal salts, particularly calcium, magnesium, sodium, lithium, zinc, potassium, and iron salts. Compounds included in the present compositions that include a basic or acidic moiety may also form pharmaceutically acceptable salts with various amino acids. The compounds of the disclosure may contain both acidic and basic groups; for example, one amino and one carboxylic acid group. In such a case, the compound can exist as an acid addition salt, a zwitterion, or a base salt.
The term “therapeutically effective amount” or “effective amount” as used herein refers to the amount of the subject compound that will elicit the biological or medical response of a tissue, system or animal, (e.g., mammal or human) that is being sought by the researcher, veterinarian, medical doctor or other clinician. The compounds or pharmaceutical compositions of the disclosure are administered in therapeutically effective amounts to treat a disease. Alternatively, a therapeutically effective amount of a compound is the quantity required to achieve a desired therapeutic and/or prophylactic effect.
The term “treating” includes any effect, e.g., lessening, reducing, modulating, or eliminating, via disruption of HBV core protein assembly, that results in the improvement of the disease. “Disruption” includes inhibition of HBV viral assembly and infection.
The compounds of the disclosure may contain one or more chiral centers and, therefore, exist as stereoisomers. The term “stereoisomers” when used herein consist of all enantiomers or diastereomers. These compounds may be designated by the symbols “(+),” “(- ),” “R” or “S,” depending on the configuration of substituents around the stereogenic carbon atom, but the skilled artisan will recognize that a structure may denote a chiral center implicitly. The present disclosure encompasses various stereoisomers of these compounds and mixtures thereof. Mixtures of enantiomers or diastereomers may be designated “(+)” in nomenclature, but the skilled artisan will recognize that a structure may denote a chiral center implicitly.
The compounds of the disclosure may contain one or more double bonds and, therefore, exist as geometric isomers resulting from the arrangement of substituents around a carbon-carbon double bond. The symbol denotes a bond that may be a single, double or triple bond as described herein. Substituents around a carbon-carbon double bond are designated as being in the “Z’ or “E” configuration wherein the terms “Z’ and “E” are used in accordance with IUPAC standards. Unless otherwise specified, structures depicting double bonds encompass both the “E” and “Z” isomers. Substituents around a carbon-carbon double bond alternatively can be referred to as “cis” or “trans,” where “cis” represents substituents on the same side of the double bond and “trans” represents substituents on opposite sides of the double bond.
Compounds of the disclosure may contain a carbocyclic or heterocyclic ring and therefore, exist as geometric isomers resulting from the arrangement of substituents around the ring. The arrangement of substituents around a carbocyclic or heterocyclic ring are designated as being in the “Z” or “E” configuration wherein the terms “Z” and “E” are used in accordance with IUPAC standards. Unless otherwise specified, structures depicting carbocyclic or heterocyclic rings encompass both “Z” and “E” isomers. Substituents around a carbocyclic or heterocyclic ring may also be referred to as “cis” or “trans”, where the term “cis” represents substituents on the same side of the plane of the ring and the term “trans” represents substituents on opposite sides of the plane of the ring. Mixtures of compounds wherein the substituents are disposed on both the same and opposite sides of plane of the ring are designated “cis/trans.”
Individual enantiomers and diastereomers of compounds of the present disclosure can be prepared synthetically from commercially available starting materials that contain asymmetric or stereogenic centers, or by preparation of racemic mixtures followed by resolution methods well known to those of ordinary skill in the art. These methods of resolution are exemplified by (1) attachment of a mixture of enantiomers to a chiral auxiliary, separation of the resulting mixture of diastereomers by recrystallization or chromatography and liberation of the optically pure product from the auxiliary, (2) salt formation employing an optically active resolving agent, (3) direct separation of the mixture of optical enantiomers on chiral liquid chromatographic columns or (4) kinetic resolution using stereoselective chemical or enzymatic reagents. Racemic mixtures can also be resolved into their component enantiomers by well-known methods, such as chiral-phase liquid chromatography or crystallizing the compound in a chiral solvent. Stereoselective syntheses, a chemical or enzymatic reaction in which a single reactant forms an unequal mixture of stereoisomers during the creation of a new stereocenter or during the transformation of a pre-existing one, are well known in the art. Stereoselective syntheses encompass both enantiomeric and diastereoselective transformations and may involve the use of chiral auxiliaries. For examples, see Carreira and Kvaemo, Classics in Stereoselective Synthesis, Wiley-VCH: Weinheim, 2009.
The compounds disclosed herein can exist in solvated as well as unsolvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is intended that the disclosure embrace both solvated and unsolvated forms. In one embodiment, the compound is amorphous. In one embodiment, the compound is a single polymorph. In another embodiment, the compound is a mixture of polymorphs. In another embodiment, the compound is in a crystalline form.
The disclosure also embraces isotopically labeled compounds of the disclosure which are identical to those recited herein, except that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into compounds of the disclosure include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine and chlorine, such as 2H, 3H, 13C, 14C, 15N, 18O, 170, 31P, 32P, 35S, 18F, and 36C1, respectively. For example, a compound of the disclosure may have one or more H atom replaced with deuterium.
Certain isotopically-labeled disclosed compounds (e.g., those labeled with 3H and 14C) are useful in compound and/or substrate tissue distribution assays. Tritiated (i.e., 3H) and
carbon-14 (i.e., 14C) isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium (i.e., 2H) may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances. Isotopically labeled compounds of the disclosure can generally be prepared by following procedures analogous to those disclosed in the examples herein by substituting an isotopically labeled reagent for a non-isotopically labeled reagent.
The term “prodrug” refers to compounds that are transformed in vivo to yield a disclosed compound or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (such as by esterase, amidase, phosphatase, oxidative and or reductive metabolism) in various locations (such as in the intestinal lumen or upon transit of the intestine, blood or liver). Prodrugs are well known in the art (for example, see Rautio, Kumpulainen, et al., Nature Reviews Drug Discovery 2008, 7, 255).
II. 5-Membered Heteroaryl Carboxamide Compounds
In one aspect, the present disclosure provides a compound of Formula I R2
 Formula I
Formula I
, or a pharmaceutically acceptable salt thereof, wherein:
L1 is a bond, Ci-4alkylene, Ci-4alkenylene, Ci-4alkynylene, haloCi-4alkylene, hydroxyCi-4alkylene, NRcCi-4alkyl, OC alkyl, O, NRC, C(O), C(O)O, C(O)NRC, S(O)t, S(O)tNRc, S(O)tCi-4alkyl, and S(O)thaloCi-4alkyl;
L3 is Ci-ealkylene, C2-ealkenylene or C2-ealkynylene, wherein the Ci-ealkylene, C2- ealkenylene, C2-ealkynylene is optionally substituted with 1-10 substituents independently selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci- ealkoxy, haloCi-ealkoxy, hydroxyCi-ealkoxy, RaRbN-Ci-ealkoxy, and haloCi-ealkylNR0-;
X1 is NRX1, O or S;
X4 is O or S;
X5 is O, S or NR6a;
Ra, Rb and Rc are independently selected for each occurrence from the group consisting of hydrogen, Ci-6 alkyl, and haloCi -ealkyl;
Rd is hydrogen, OH, Ci-6 alkyl or Ci-ealkoxy;
Rxl is hydrogen, Ci-4 alkyl, Ci-4 alkenyl, Ci-4 alkynyl, haloCi-4 alkyl, or C3-6 monocycloalkyl;
ROa is independently selected for each occurrence from the group consisting of hydrogen, halogen, OH, CN, NO2, RaRbN-, Ci-4alkyl and haloCi-4 alkyl;
R6a is hydrogen, C1-4 alkyl, haloCi-4 alkyl or C3-4cycloalkyl;
R6b is Ci-ealkyl, C2-ealkenyl or C2-ealkynyl, wherein the Ci-ealkyl, C2-ealkenyl, C2- ealkynyl is optionally substituted with 1-10 substituents independently selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi-ealkoxy, RaRbN-Ci-ealkoxy, and haloCi-ealkylNFC-;
R°, R4a, R6 and R11 are independently selected for each occurrence from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, R6b, R6bC(O)-, R6bC(O)O-, R6bC(O)NRc-, R6bS(O)tNRc-, R6bS(O)t-, R6bO-, R6bNRc-, R6bC(O)-L3-, and R6bC(O)O-L3-, R6bC(O)NRc-L3-, R6bS(O)tNRc-L3-, R6bS(O)q-L3-, R6bO-L3-, and R6bNRc-L3-;
R1 is a phenyl or 5-6 membered monocyclic heteroaryl, wherein the phenyl or 5-6 membered monocyclic heteroaryl is optionally substituted with one, two, or three independently selected R11 groups;
R2 and R8 are independently selected from the group consisting of hydrogen, halo, CN, OH, RaRbN, Ci-4alkyl, haloCi^alkyl, Cs-smonocycloalkyl, Ci-4alkoxy, and haloCi- 4alkoxy;



R5d is selected from the group consisting of:
 p is independently selected for each occurrence from the group consisting of 0, 1, 2 and 3; r is independently selected for each occurrence from the group consisting of 0, 1 and
p is independently selected for each occurrence from the group consisting of 0, 1, 2 and 3; r is independently selected for each occurrence from the group consisting of 0, 1 and
2; t is independently selected for each occurrence from the group consisting of 0, 1 and 2; v is independently selected for each occurrence from the group consisting of 0, 1, 2 and 3; and
w is independently selected for each occurrence from the group consisting of 0, 1 and
2.
The following embodiments further describe a compound of Formula I, or a pharmaceutically acceptable salt thereof.
In certain embodiments, X1 is S.
In certain embodiments, X1 is NRxl.
In certain embodiments, X1 is NRxl and Rxl is hydrogen of methyl.
In certain embodiments, X1 is NRxl and Rxl is methyl.
In certain embodiments, L1 is a bond.
In certain embodiments, L1 is Ci^alkylene.
In certain embodiments, p is 0.
In certain embodiments, R° is selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN- , RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-6alkylS(O)t-, Ci-6alkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi- ealkoxy-, RaRbNCi-ealkoxy-, RaRbNCi-6alkylNRc-, Ci-6alkylNRaCi-6alkyleneNRc-, Ci- ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-6alkylS(O)tCi- ealkylene-, Ci-6alkylS(O)tNRaCi-6alkylene-, Ci-6alkylC(O)Ci-6alkylene-, Ci-6alkylC(O)OCi- ealkylene- and R9, wherein:
R9 is R12S(O)t-Ci-6alkylene-, R12S(O)tNH-Ci-6alkylene-, R12C(O)NH-Ci-6alkylene-, R12S(O)t-haloCi-6alkylene-, R12S(O)tNH-haloCi-6alkylene-, or R12C(O)NH-haloCi-6alkylene-; and
R12 is RaRbN-, Ci-ealkyl, Ci-ehaloalkyl, Ci-ealkoxy, or Ci-ehaloalkoxy.
In certain embodiments, R° is selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN- , RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-ealkylS(O)t-, Ci-ealkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi- ealkoxy-, RaRbNCi-ealkoxy-, RaRbNCi-6alkylNRc-, Ci-6alkylNRaCi-6alkyleneNRc-, Ci- ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-ealkylS(O)tCi- ealkylene-, Ci-6alkylS(O)tNRaCi-ealkylene-, Ci-6alkylC(O)Ci-6alkylene-, and Ci- 6alkylC(O)OCi-6alkylene-.
In certain embodiments, R° is R9; wherein:
R9 is R12S(O)t-Ci-6alkylene-, R12S(O)tNH-Ci-6alkylene-, R12C(O)NH-Ci-6alkylene-, R12S(O)t-haloCi-6alkylene-, R12S(O)tNH-haloCi-6alkylene-, or R12C(O)NH-haloCi-6alkylene-; and
R12 is RaRbN-, Ci-ealkyl, Ci-ehaloalkyl, Ci-ealkoxy, or Ci-ehaloalkoxy.
T J-(R1 1)zi
In certain embodiments, R1 is ; R11 is independently selected for each occurrence from the group consisting of halogen, CN, Ci-ealkyl and haloCi -ealkyl; and zl is 0, 1, 2 or 3.
In certain embodiments, R11 is independently selected for each occurrence from the group consisting of halogen and CN.
In certain embodiments, R11 is independently selected for each occurrence from the group consisting of F, Cl, Br and I.
In certain embodiments, R1 is selected from the group consisting of:

In certain embodiments,

In certain embodiments, X1 is NRxl, Rxl is hydrogen or methyl, and R1 is

In certain embodiments, R2 is hydrogen.
In certain embodiments, X1 is NRxl, Rxl is hydrogen or methyl, R1 is
 and R2 is hydrogen.
and R2 is hydrogen.
In certain embodiments,
 In certain embodiments,
In certain embodiments,

In certain embodiments, R3 is
 .
.
In certain embodiments,


In certain embodiments,
 In certain embodiments,
In certain embodiments,

In certain embodiments,
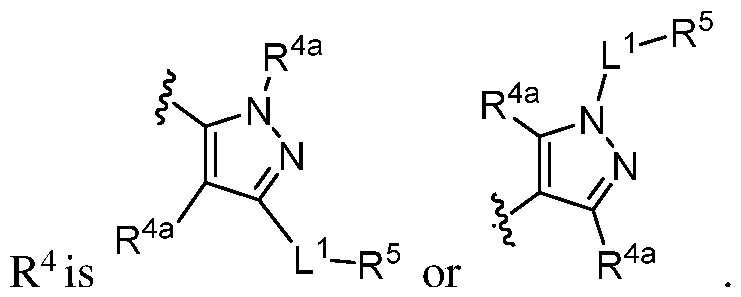
In certain embodiments,
 wherein:
wherein:
R4b is selected for each occurrence from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-6alkylS(O)qt-, Ci-ealkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi -ealkoxy-, RaRbNCi-ealkoxy-, RaRbNCi-ealkylNRc-, Ci-ealkylNRaCi-ealkyleneNRc-, Ci-ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-ealkylS(O)tCi- ealkylene-, Ci-ealkylS(O)tNRaCi-ealkylene-, Ci-6alkylC(O)Ci-ealkylene-, Ci-ealkylC(O)OCi- ealkylene- and R9, wherein:
R9 is R12S(O)t-Ci-6alkylene-, R12S(O)tNH-Ci-6alkylene-, R12C(O)NH-Ci-6alkylene-, R12S(O)t-haloCi-6alkylene-, R12S(O)tNH-haloCi-6alkylene-, or R12C(O)NH-haloCi-6alkylene-; and
R12 is RaRbN-, Ci-ealkyl, Ci-ehaloalkyl, Ci-ealkoxy, or Ci-ehaloalkoxy.
In certain embodiments,
 wherein:
R4b is selected for each occurrence from the group consisting of \ hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazine, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-ealkylS(O)t-, Ci-ealkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi- ealkoxy-, RaRbNCi-6alkoxy-, RaRbNCi-6alkylNRc-, Ci-6alkylNRaCi-6alkyleneNRc-, Ci- ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-ealkylS(O)tCi- ealkylene-, Ci-6alkylS(O)tNRaCi-ealkylene-, Ci-6alkylC(O)Ci-6alkylene-, and Ci- 6alkylC(O)OCi-6alkylene-.
wherein:
R4b is selected for each occurrence from the group consisting of \ hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazine, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-ealkylS(O)t-, Ci-ealkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi- ealkoxy-, RaRbNCi-6alkoxy-, RaRbNCi-6alkylNRc-, Ci-6alkylNRaCi-6alkyleneNRc-, Ci- ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-ealkylS(O)tCi- ealkylene-, Ci-6alkylS(O)tNRaCi-ealkylene-, Ci-6alkylC(O)Ci-6alkylene-, and Ci- 6alkylC(O)OCi-6alkylene-.
In certain embodiments,

In certain embodiments,

In certain embodiments, R5 is R5a, R5b, R5d or R6.
In certain embodiments, R5 is R5a, R5d or R6.
In certain embodiments, R5 is R5a or R5d.
In certain embodiments, R4 is R6.
In certain embodiments, R4 is R5a.
In certain embodiments, R4 is R5d.
In certain embodiments, L1 is a bend, Ci-4alkylene, Ci-4alkenylene, Ci^alkynylene, haloCi-4alkylene or hydroxyCi-4alkylene.
In certain embodiments, L1 is a bond.
In certain embodiments, R6 is selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN- , RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-6alkylS(O)t-, Ci-6alkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi- ealkoxy-, RaRbNCi-ealkoxy-, RaRbNCi-6alkylNRc-, Ci-6alkylNRaCi-6alkyleneNRc-, Ci- ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-6alkylS(O)tCi- ealkylene-, Ci-6alkylS(O)tNRaCi-6alkylene-, Ci-6alkylC(O)Ci-6alkylene-, Ci-6alkylC(O)OCi- ealkylene- and R9, wherein:
R9 is R12S(O)t-Ci-6alkylene-, R12S(O)tNH-Ci-6alkylene-, R12C(O)NH-Ci-6alkylene-, R12S(O)t-haloCi-6alkylene-, R12S(O)tNH-haloCi-6alkylene-, or R12C(O)NH-haloCi-6alkylene-; and
R12 is RaRbN-, Ci-ealkyl, Ci-ehaloalkyl, Ci-ealkoxy, or Ci-ehaloalkoxy.
In certain embodiments, R6 is selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN- , RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-6alkylS(O)t-, Ci-6alkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi- ealkoxy-, RaRbNCi-6alkoxy-, RaRbNCi-6alkylNRc-, Ci-6alkylNRaCi-6alkyleneNRc-, Ci- ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-6alkylS(O)tCi- ealkylene-, Ci-6alkylS(O)tNRaCi-6alkylene-, Ci-6alkylC(O)Ci-6alkylene-, and Ci- 6alkylC(O)OCi-6alkylene-.
In certain embodiments, R6 is R9, wherein:
R9 is R12S(O)t-Ci-6alkylene-, R12S(O)tNH-Ci-6alkylene-, R12C(O)NH-Ci-6alkylene-, R12S(O)t-haloCi-6alkylene-, R12S(O)tNH-haloCi-6alkylene-, or R12C(O)NH-haloCi-6alkylene-; and
R12 is RaRbN-, Ci-ealkyl, Ci-ehaloalkyl, Ci-ealkoxy, or Ci-ehaloalkoxy.
In certain embodiments, R8 is hydrogen, halogen, methyl, methoxy or OH.
In certain embodiments, R8 is hydrogen or OH.
In certain embodiments, R8 is OH.
In certain embodiments, X1 is NRxl; Rxl is hydrogen or methyl; R1 is

 hydrogen, OH or Ci-ealkoxy.
hydrogen, OH or Ci-ealkoxy.
In certain embodiments, X1 is NRxl; Rxl is hydrogen or methyl; R1 is


It will be appreciated that all chemically allowable combinations of the aforementioned embodiments are also contemplated as embodiments of the invention.
III. Pharmaceutical Compositions and Kits
In another aspect, the disclosure provides pharmaceutical compositions comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. In particular, the present disclosure provides pharmaceutical compositions comprising compounds as disclosed herein formulated together with one or more pharmaceutically acceptable carriers. These formulations include those suitable for oral, rectal, topical, buccal, parenteral (e.g., subcutaneous, intramuscular, intradermal, or intravenous), rectal, vaginal, or aerosol administration, although the most suitable form of administration in any given case will depend on the degree and severity of
the condition being treated and on the nature of the particular compound being used. For example, disclosed compositions may be formulated as a unit dose, and/or may be formulated for oral or subcutaneous administration.
In another aspect, the disclosure provides a pharmaceutical composition comprises a compound according to any combination of the Examples described herein, or a pharmaceutically acceptable salt and/or stereoisomer thereof.
Exemplary pharmaceutical compositions of this disclosure may be used in the form of a pharmaceutical preparation, for example, in solid, semisolid or liquid form, which contains one or more compounds of the disclosure, as an active ingredient, in admixture with an organic or inorganic carrier or excipient suitable for external, enteral or parenteral applications. The active ingredient may be compounded, for example, with the usual nontoxic, pharmaceutically acceptable carriers for tablets, pellets, capsules, suppositories, solutions, emulsions, suspensions, and any other form suitable for use. The active object compound is included in the pharmaceutical composition in an amount sufficient to produce the desired effect upon the process or condition of the disease.
For preparing solid compositions such as tablets, the principal active ingredient may be mixed with a pharmaceutical carrier, e.g., conventional tableting ingredients such as corn starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium phosphate or gums, and other pharmaceutical diluents, e.g., water, to form a solid preformulation composition containing a homogeneous mixture of a compound of the disclosure, or a nontoxic pharmaceutically acceptable salt thereof. When referring to these preformulation compositions as homogeneous, it is meant that the active ingredient is dispersed evenly throughout the composition so that the composition may be readily subdivided into equally effective unit dosage forms such as tablets, pills and capsules.
In solid dosage forms for oral administration (capsules, tablets, pills, dragees, powders, granules and the like), the subject composition is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example, acetyl alcohol and glycerol monostearate; (8)
absorbents, such as kaolin and bentonite clay; (9) lubricants, such a talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof; and (10) coloring agents. In the case of capsules, tablets and pills, the compositions may also comprise buffering agents. Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
A tablet may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface-active or dispersing agent. Molded tablets may be made by molding in a suitable machine a mixture of the subject composition moistened with an inert liquid diluent. Tablets, and other solid dosage forms, such as dragees, capsules, pills and granules, may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art.
Compositions for inhalation or insufflation include solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders. Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs. In addition to the subject composition, the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, cyclodextrins and mixtures thereof.
Suspensions, in addition to the subject composition, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
Formulations for rectal or vaginal administration may be presented as a suppository, which may be prepared by mixing a subject composition with one or more suitable nonirritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the body cavity and release the active agent.
Dosage forms for transdermal administration of a subject composition include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants. The active component may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants which may be required.
The ointments, pastes, creams and gels may contain, in addition to a subject composition, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
Powders and sprays may contain, in addition to a subject composition, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances. Sprays may additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
Compositions and compounds of the present disclosure may alternatively be administered by aerosol. This is accomplished by preparing an aqueous aerosol, liposomal preparation or solid particles containing the compound. A non-aqueous (e.g., fluorocarbon propellant) suspension could be used. Sonic nebulizers may be used because they minimize exposing the agent to shear, which may result in degradation of the compounds contained in the subject compositions. Ordinarily, an aqueous aerosol is made by formulating an aqueous solution or suspension of a subject composition together with conventional pharmaceutically acceptable carriers and stabilizers. The carriers and stabilizers vary with the requirements of the particular subject composition, but typically include non-ionic surfactants (Tweens, Pluronics, or polyethylene glycol), innocuous proteins like serum albumin, sorbitan esters, oleic acid, lecithin, amino acids such as glycine, buffers, salts, sugars or sugar alcohols. Aerosols generally are prepared from isotonic solutions.
Pharmaceutical compositions of this disclosure suitable for parenteral administration comprise a subject composition in combination with one or more pharmaceutically- acceptable sterile isotonic aqueous or non-aqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
Examples of suitable aqueous and non-aqueous carriers which may be employed in the pharmaceutical compositions of the disclosure include water, ethanol, polyols (such as
glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate and cyclodextrins. Proper fluidity may be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
In another aspect, the disclosure provides enteral pharmaceutical formulations including a disclosed compound and an enteric material; and a pharmaceutically acceptable carrier or excipient thereof. Enteric materials refer to polymers that are substantially insoluble in the acidic environment of the stomach, and that are predominantly soluble in intestinal fluids at specific pHs. The small intestine is the part of the gastrointestinal tract (gut) between the stomach and the large intestine, and includes the duodenum, jejunum, and ileum. The pH of the duodenum is about 5.5, the pH of the jejunum is about 6.5 and the pH of the distal ileum is about 7.5. Accordingly, enteric materials are not soluble, for example, until a pH of about 5.0, of about 5.2, of about 5.4, of about 5.6, of about 5.8, of about 6.0, of about 6.2, of about 6.4, of about 6.6, of about 6.8, of about 7.0, of about 7.2, of about 7.4, of about 7.6, of about 7.8, of about 8.0, of about 8.2, of about 8.4, of about 8.6, of about 8.8, of about 9.0, of about 9.2, of about 9.4, of about 9.6, of about 9.8, or of about 10.0. Exemplary enteric materials include cellulose acetate phthalate (CAP), hydroxypropyl methylcellulose phthalate (HPMCP), polyvinyl acetate phthalate (PVAP), hydroxypropyl methylcellulose acetate succinate (HPMCAS), cellulose acetate trimellitate, hydroxypropyl methylcellulose succinate, cellulose acetate succinate, cellulose acetate hexahydrophthalate, cellulose propionate phthalate, cellulose acetate maleate, cellulose acetate butyrate, cellulose acetate propionate, copolymer of methylmethacrylic acid and methyl methacrylate, copolymer of methyl acrylate, methylmethacrylate and methacrylic acid, copolymer of methylvinyl ether and maleic anhydride (Gantrez ES series), ethyl methyacrylate-methylmethacrylate- chlorotrimethylammonium ethyl acrylate copolymer, natural resins such as zein, shellac and copal collophorium, and several commercially available enteric dispersion systems (e. g. , Eudragit L30D55, Eudragit FS30D, Eudragit L100, Eudragit S100, Kollicoat EMM30D, Estacryl 30D, Coateric, and Aquateric). The solubility of each of the above materials is either known or is readily determinable in vitro. The foregoing is a list of possible materials, but one of skill in the art with the benefit of the disclosure would recognize that it is not comprehensive and that there are other enteric materials that would meet the objectives of the present disclosure.
Advantageously, the disclosure also provides kits for use by e.g., a consumer in need of HBV infection treatment. Such kits include a suitable dosage form such as those described above and instructions describing the method of using such dosage form tomediate, reduce or prevent HBV infection. The instructions would direct the consumer or medical personnel to administer the dosage form according to administration modes known to those skilled in the art. Such kits could advantageously be packaged and sold in single or multiple kit units. An example of such a kit is a so-called blister pack. Blister packs are well known in the packaging industry and are being widely used for the packaging of pharmaceutical unit dosage forms (tablets, capsules, and the like). Blister packs generally consist of a sheet of relatively stiff material covered with a foil of a preferably transparent plastic material. During the packaging process recesses are formed in the plastic foil. The recesses have the size and shape of the tablets or capsules to be packed. Next, the tablets or capsules are placed in the recesses and the sheet of relatively stiff material is sealed against the plastic foil at the face of the foil which is opposite from the direction in which the recesses were formed. As a result, the tablets or capsules are sealed in the recesses between the plastic foil and the sheet. Preferably the strength of the sheet is such that the tablets or capsules can be removed from the blister pack by manually applying pressure on the recesses whereby an opening is formed in the sheet at the place of the recess. The tablet or capsule can then be removed via said opening.
It may be desirable to provide a memory aid on the kit, e.g., in the form of numbers next to the tablets or capsules whereby the numbers correspond with the days of the regimen which the tablets or capsules so specified should be ingested. Another example of such a memory aid is a calendar printed on the card, e.g., as follows “First Week, Monday, Tuesday, . . . etc. . . . Second Week, Monday, Tuesday, . . .” etc. Other variations of memory aids will be readily apparent. A “daily dose” can be a single tablet or capsule or several pills or capsules to be taken on a given day. Also, a daily dose of a first compound can consist of one tablet or capsule while a daily dose of the second compound can consist of several tablets or capsules and vice versa. The memory aid should reflect this.
IV. Methods
In a further aspect, a method for treating a hepatitis B infection in a patient in need thereof is provided, comprising administering to a subject or patient an effective amount of a disclosed compound, and/or administering a first disclosed compound and optionally, an additional, different disclosed compound(s). In another embodiment, a method for treating a
hepatitis B infection in a patient in need thereof is provided, comprising administering to a subject or patient a therapeutically effective amount of a disclosed pharmaceutical composition or a pharmaceutical composition comprising a disclosed compound, or two or more disclosed compounds, and a pharmaceutically acceptable excipient.
For use in accordance with this aspect, the appropriate dosage is expected to vary depending on, for example, the particular compound employed, the mode of administration, and the nature and severity of the infection to be treated as well as the specific infection to be treated and is within the purview of the treating physician. Usually, an indicated administration dose may be in the range between about 0.1 to about 1000 pg/kg body weight. In some cases, the administration dose of the compound may be less than 400 pg/kg body weight. In other cases, the administration dose may be less than 200 pg/kg body weight. In yet other cases, the administration dose may be in the range between about 0.1 to about 100 pg/kg body weight. The dose may be conveniently administered once daily, or in divided doses up to, for example, four times a day or in sustained release form.
A compound of the present disclosure may be administered by any conventional route, in particular: enterally, topically, orally, nasally, e.g., in the form of tablets or capsules, via suppositories, or parenterally, e.g., in the form of injectable solutions or suspensions, for intravenous, intra-muscular, sub-cutaneous, or intra-peritoneal injection. Suitable formulations and pharmaceutical compositions will include those formulated in a conventional manner using one or more physiologically acceptable carriers or excipients, and any of those known and commercially available and currently employed in the clinical setting. Thus, the compounds may be formulated for oral, buccal, topical, parenteral, rectal or transdermal administration or in a form suitable for administration by inhalation or insufflation (either orally or nasally).
For oral administration, pharmaceutical compositions may take the form of, for example, tablets or capsules prepared by conventional means with pharmaceutically acceptable excipients such as binding agents (e.g., pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g. lactose, microcrystalline cellulose or calcium hydrogen phosphate); lubricants (e.g., magnesium stearate, talc or silica); disintegrants (e.g., potato starch or sodium starch glycollate); or wetting agents (e.g., sodium lauryl sulphate). Tablets may be coated by methods well known in the art. Liquid preparations for oral administration may take the form of, for example, solutions, syrups or suspensions, or they may be presented as a dry product for constitution with water or other suitable vehicle before use. Such liquid preparations may be prepared by conventional means
with pharmaceutically acceptable additives such as suspending agents (e.g., sorbitol syrup, cellulose derivatives or hydrogenated edible fats); emulsifying agents (e.g., lecithin or acacia); non-aqueous vehicles (e.g., almond oil, oily esters, ethyl alcohol or fractionated vegetable oils); and preservatives (e.g., methyl or propyl-p-hydroxybenzoates or sorbic acid). Preparations may also contain buffer salts, flavoring, coloring and sweetening agents as appropriate.
Preparations for oral administration may also be suitably formulated to give controlled-release or sustained release of the active compound(s) over an extended period. For buccal administration the compositions may take the form of tablets or lozenges formulated in a conventional manner known to the skilled artisan.
A disclosed compound may also be formulated for parenteral administration by injection e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form e.g., in ampoules or in multi-dose containers, with an added preservative. The compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain additives such as suspending, stabilizing and/or dispersing agents. Alternatively, the compound may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen- free water, before use. Compounds may also be formulated for rectal administration as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
Also contemplated herein are methods and compositions that include a second active agent or administering a second active agent. For example, in addition to being infected with HBV, a subject or patient can further have HBV infection-related co-morbidities, i.e., diseases and other adverse health conditions associated with, exacerbated by, or precipitated by being infected with HBV. Contemplated herein are disclosed compounds in combination with at least one other agent that has previously been shown to treat these HBV-infection- related conditions.
In some cases, a disclosed compound may be administered as part of a combination therapy in conjunction with one or more antivirals. Example antivirals include nucleoside analogs, interferon a, and other assembly effectors, for instance heteroaryldihydropyrimidines (HAPs) such as methyl 4-(2-chloro-4-fluorophenyl)-6-methyl- 2-(pyridin-2-yl)-l,4-dihydropyrimidine-5-carboxylate (HAP-1). For example, provided herein is a method of treating a patient suffering from hepatitis B infection comprising administering to the patient a first amount of a disclosed compound and a second amount of an antiviral, or other anti HBV agent, for example a second amount of a second compound
selected from the group consisting of: an HBV capsid assembly promoter (for example,
GES4, BAY 41-4109, AT-130, DVR-23 (e.g., as depicted below),

NVR 3-778, NVR1221 (by code); and N890 (as depicted below):
 other capsid inhibitors such as those disclosed in the following patent applications hereby incorporated by reference: W02014037480, WO2014184328, W02013006394, WO2014089296, W02014106019, WO2013102655, WO2014184350, WO2014184365, WO2014161888, WO2014131847, WO2014033176, WO2014033167, and W02014033170; Nucleos(t)ide analogs interfering with viral polymerase, such as entecavir (Baraclude), Lamivudine, (Epivir-HBV), Telbivudine (Tyzeka, Sebivo), Adefovir dipivoxil (Hepsera), Tenofovir (Viread), Tenofovir alafenamide fumarate (TAF), prodrugs of tenofavir (e.g. AGX-1009), L-FMAU (Clevudine), EB80380 (Besifovir) and:
other capsid inhibitors such as those disclosed in the following patent applications hereby incorporated by reference: W02014037480, WO2014184328, W02013006394, WO2014089296, W02014106019, WO2013102655, WO2014184350, WO2014184365, WO2014161888, WO2014131847, WO2014033176, WO2014033167, and W02014033170; Nucleos(t)ide analogs interfering with viral polymerase, such as entecavir (Baraclude), Lamivudine, (Epivir-HBV), Telbivudine (Tyzeka, Sebivo), Adefovir dipivoxil (Hepsera), Tenofovir (Viread), Tenofovir alafenamide fumarate (TAF), prodrugs of tenofavir (e.g. AGX-1009), L-FMAU (Clevudine), EB80380 (Besifovir) and:
 viral entry inhibitors such as Myrcludex B and related lipopeptide derivatives; HBsAg secretion inhibitors such as REP 9AC’ and related nucleic acid-based amphipathic polymers, HBF-0529 (PBHBV-001), PBHBV-2-15 as depicted below:
viral entry inhibitors such as Myrcludex B and related lipopeptide derivatives; HBsAg secretion inhibitors such as REP 9AC’ and related nucleic acid-based amphipathic polymers, HBF-0529 (PBHBV-001), PBHBV-2-15 as depicted below:
 and BM601 as depicted below:
and BM601 as depicted below:
 disruptors of nucleocapsid formation or integrity such as NZ-4/W28F:
disruptors of nucleocapsid formation or integrity such as NZ-4/W28F:
 cccDNA formation inhibitors such as BSBI-25, CCC-0346, CCC-0975 (as depicted below):
cccDNA formation inhibitors such as BSBI-25, CCC-0346, CCC-0975 (as depicted below):

HBc directed transbodies such as those described in Wang Y, et al, Transbody against hepatitis B virus core protein inhibits hepatitis B virus replication in vitro, Int.
Immunopharmacol (2014), located at //dx.doi.org/10.1016/j.intimp.2015.01.028; antiviral core protein mutant (such as Cpl83-V124W and related mutations as described in WO/2013/010069, W02014/074906, each incorporated by reference); inhibitors of HBx- interactions such as RNAi, antisense and nucleic acid based polymers targeting HBV RNA;, e.g., RNAi (for example ALN-HBV, ARC-520, TKM-HBV, ddRNAi), antisense (ISIS- HBV), or nucleic acid based polymer: (REP 2139-Ca); immunostimulants such as Interferon alpha 2a (Roferon), Intron A (interferon alpha 2b), Pegasys (peginterferon alpha 2a), Pegylated IFN 2b, IFN lambda la and PEG IFN lambda la, Wellferon, Roferon, Infergen, lymphotoxin beta agonists such as CBE11 and BS1); Non- Interferon Immune enhancers such as Thymosin alpha- 1 (Zadaxin) and Interleukin-7 (CYT107); TER-7/9 agonists such as GS- 9620, CYT003, Resiquimod; Cyclophilin inhibitors such as NVP018; GCB-030; SCY-635;
Alisporivir; NIM811 and related cyclosporine analogs; vaccines such as GS-4774, TG1050, Core antigen vaccine; SMAC mimetics such as birinapant and other IAP- antagonists; Epigenetic modulators such as KMT inhibitors (EZH1/2, G9a, SETD7, Suv39 inhibitors), PRMT inhibitors, HDAC inhibitors, SIRT agonists, HAT inhibitors, WD antagonists (e.g. OICR-9429), PARP inhibitors, APE inhibitors, DNMT inhibitors, LSD1 inhibitors, JMJD HDM inhibitors, and Bromodomain antagonists; kinase inhibitors such as TKB1 antagonists, PLK1 inhibitors, SRPK inhibitors, CDK2 inhibitors, ATM & ATR kinase inhibitors; STING Agonists; Ribavirin; N-acetyl cysteine ; NOV-205 (BAM205); Nitazoxanide (Alinia), Tizoxanide; SB 9200 Small Molecule Nucleic Acid Hybrid (SMNH); DV-601; Arbidol; FXR agonists (such as GW 4064 and Fexaramin); antibodies, therapeutic proteins, gene therapy, and biologies directed against viral components or interacting host proteins.
In some embodiments, the disclosure provides a method of treating a hepatitis B infection in a patient in need thereof, comprising administering a first compound selected from any one of the disclosed compounds, and one or more other HBV agents each selected from the group consisting of HBV capsid assembly promoters, HBF viral polymerase interfering nucleosides, viral entry inhibitors, HBsAg secretion inhibitors, disruptors of nucleocapsid formation, cccDNA formation inhibitors, antiviral core protein mutant, HBc directed transbodies, RNAi targeting HBV RNA, immunostimulants, TLR-7/9 agonists, cyclophilin inhibitors, HBV vaccines, SMAC mimetics, epigenetic modulators, kinase inhibitors, and STING agonists. In some embodiments, the disclosure provides a method of treating a hepatitis B infection in a patient in need thereof, comprising administering an amount of a disclosed compound, and administering another HBV capsid assembly promoter.
In some embodiments, the first and second amounts together comprise a pharmaceutically effective amount. The first amount, the second amount, or both may be the same, more, or less than effective amounts of each compound administered as monotherapies. Therapeutically effective amounts of a disclosed compound and antiviral may be coadministered to the subject, i.e., administered to the subject simultaneously or separately, in any given order and by the same or different routes of administration. In some instances, it may be advantageous to initiate administration of a disclosed compound first, for example one or more days or weeks prior to initiation of administration of the antiviral. Moreover, additional drugs may be given in conjunction with the above combination therapy.
In another embodiment, a disclosed compound may be conjugated (e.g., covalently bound directly or through molecular linker to a free carbon, nitrogen (e.g., an amino group), or oxygen (e.g., an active ester) of a disclosed compound), with a detection moiety, for e.g., a
fluorophore moiety (such a moiety may for example re-emit a certain light frequency upon binding to a virus and/or upon photon excitation). Contemplated fluorophores include AlexaFluor® 488 (Invitrogen) and BODIPY FL (Invitrogen), as well as fluorescein, rhodamine, cyanine, indocarbocyanine, anthraquinones, fluorescent proteins, aminocoumarin, methoxycoumarin, hydroxycoumarin, Cy2, Cy3, and the like. Such disclosed compounds conjugated to a detection moiety may be used in e.g., a method for detecting HBV or biological pathways of HBV infection, e.g., in vitro or in vivo; and/or methods of assessing new compounds for biological activity.
V. Examples
The compounds described herein can be prepared in a number of ways based on the teachings contained herein and synthetic procedures known in the art. In the description of the synthetic methods described below, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and workup procedures, can be chosen to be the conditions standard for that reaction, unless otherwise indicated. It is understood by one skilled in the art of organic synthesis that the functionality present on various portions of the molecule should be compatible with the reagents and reactions proposed. Substituents not compatible with the reaction conditions will be apparent to one skilled in the art, and alternate methods are therefore indicated. The starting materials for the examples are either commercially available or are readily prepared by standard methods from known materials.
At least some of the compounds identified as “intermediates” herein are contemplated as compounds of the disclosure.
Abbreviations:
AcOH Acetic acid
ACN Acetonitrile
BOC2O Di-tert-butyl dicarbonate nBuLi n-Butyllithium
DCM Dichloromethane
DIAD Diisopropyl azodicarboxylate
DIEADiisopropyl ethylamine DMF N,N-Dimethylformamide DMSO Dimethyl sulfoxide
DPPF 1,1’ -Bis(diphenylphosphino)ferrocene
EA, EtOAc Ethyl acetate
El N Triethylamine
HATU Hexafluorophosphate Azabenzotriazole Tetramethyl Uronium h, hr Hour(s)
HPLC High performance liquid chromatography
LCMS Liquid chromatography-mass spectrometry
MeOH Methanol
NMON-Methylmorpholine-N-Oxide
NBS N-Bromosuccinimide
PE Petroleum ether iPrOH Isopropanol rt, r.t.Room temperature
SFC Supercritical Fluid Chromatography
TEA Triethylamine
TFA Trifluoroacetic acid
THF Tetrahydrofuran
TEC Thin-layer chromatography
XPhos 2-Dicyclohexylphosphino-2’ ,4’ ,6’-triisopropylbiphenyl
Scheme I

The synthesis of the common intermediate 1-9 is illustrated in Scheme I. The bicyclic octadien, 1-1) can be converted to the corresponding boranae ester 1-3 in two steps. Likewise, The the imidazole bromide intermediate can be synthesized starting from imidazole ester I- 4in three steps. The two compounds, 1-3 and 1-7 can be coupled together via a palladium catalyzed coupling reaction to prived 1-8. Reductionof the double bound can be carried out using standard Pd or Pt catalyzed hydrogenation reaction conditions.
Scheme II

An alternative synthesis of intermedaite 1-7 is shown in Scheme II. The method starts with imidazole carboxylic acid II which is sequentially converted to amide II-2, bis-bromated follwoed by selective debromination to removethe bromineat C-2.
Scheme III

Compounds having general structure III- 3 can be synthesized according to the methods shown in Scheme III. In the first procedure intermediate III- 1 can be treated with LDA, BuLi then reacted with 1-9 to yield III-3. In the second, C5 -halogenated (Br or I) pyrazole III- 2 undergoes metal halogen exchange with Mg, a Grignard reagent, Li or BuLi and the resulting anion reacted with 1-9 to form III-3. For R2 is Br or I, than III- 3 can be treated with boronic acid or ester, III-4 under Suzuki reaction conditions yield III-5. Likewise, compounds III- 7 and III-8 can obtained by treating III-3 (R2 = Br or I)with an amine or alkyne under Buchwald or Sonogashira reaction conditions.
Scheme IV

Scheme IV illustrates the synthesis of compounds of the general structures IV-2 and IV-5. C4 halogenated intermediate IV-1 or IV-3 can be treated with Mg, Grignard reagent, Li or BuLi to genrate the coresponding metallated hetereocycles, whch upon addtion of 1-9 form either IV-2 and IV-4, respectively. IV-4 can be further derivatized with and appropriate alkylating agent for form IV-2 and IV-5.

An alternative synthesis of IV-4 is shown in Scheme V, the nitrogen atom of the pyrazole is protected as it’ s corresponding pyrrolidine aminal.
Scheme VI

In Scheme VI an ester of propionic acid can be reacted with 1-9 under basic conditions to form VI- 1. The alkyne undergoes a reaction with an appropriately substituted hydrazine to form VI-2. This compound can be fluorinated under standard conditions to yield VI. Both VI-2 and VI-3 are further derivatized via alkylation of the pyrazole OH forming VI- 4 and VI-5, respectively.
Scheme VII

Coumpounds of general structure VII-3 and VII-5 can be synthesized according to the method shown in Scheme VII. 1-9 can be reached with an appropriately substituted 3-nitro pyrazole to form VII- 1 which can be reduced to yield VII-2. Amidation of VII-2 provide VII- 3. Halogentation of VII-2 yields VII-4, which can likewise be amidated obtain VII-5.
Following LCMS method have been used for the analysis of some of the final compounds Method A: X-Bridge BEH C-18 (3 x 50 mm x 2.5pm); Mobile phase: A; 0.025% formic acid in H2O; B; CH3CN; injection volume:2 pL; Flow rate:1.2 mL/min, column temperature: 50 °C; Gradient program: 2% B to 98% B in 2.2 min, hold until 3 min, at 3.2 min B cone, is 2 % until 4 min.
Method B: X-select CSH 18 (3 x 50 mm x 2.5pm); Mobile phase: A; 0.025% formic acid in H2O; B; CH3CN; Injection voloume:2 uL; Flow rate: 1.2 mL/min, column temperature: 50 °C; Gradient program: 0% B to 98% B in 2 min, hold until 3 min, at 3.2 min B cone, is 0 % until 4 min.
Method C: X-select CSH 18 (3 x 50mm x 2.5pm); Mobile phase: A; 0.05% formic acid in H2O:CH3CN (95:5); B; 0.05% formic acid in CH3CN; Injection volume: 2 pL; Flow rate: 1.2 mL/min, column temperature: 50 °C; Gradient program: 0% B to 98% B in 2 min, hold till 3 min, at 3.2 min B cone, is 0 % until 4 min.
Method D: X-select CSH C18 (3 x 50 mm x 2.5pm); Mobile phase: A; 2mM in Ammonium Bicarbonate; B; CH3CN; Injection voloume:2 pL; Flow rate: 1.2 mL/min, column temperature: 50 °C; Gradient program: 0% B to 98% B in 2 min, hold until 3 min, at 3.2 min B cone, is 0 % until 4 min.
Method E: X-select CSH 18 (3 x 50 mm x 2.5pm); Mobile phase: A; 0.05% formic acid in H2O; B; CH3CN; Injection volume: 2 pL; Flow rate:1.5 mL/min, column temperature: 50 °C; Gradient program: 0% B to 100% B in 1.5 min, hold until 2.2 min, at 2.6 min B cone, is 0 % until 3 min.
Synthetic procedures
Details useful for the methods described in Schemes I- VII and for the procedures that follow are listed below.
Procedure for O-Alkylation
Method 1 (Using alkyl halide)
To a stirred solution of Het-Ar-OH compound (1 eq.) and corresponding alkyl halide compound (2 eq.) in acetonitrile/DMF (4 mL/mmol) was added K2CO3 or CS2CO3 (2 eq.) and KI (0.5 eq.). The reaction mixture was stirred at 60 °C-80 °C for 2h-16 h. The reaction progress was monitored by TLC. After completion, reaction mixture was diluted with water and extracted with ethyl acetate. The combined organic layers were collected, dried over anhydrous sodium sulphate, and concentrated under reduced pressure to obtain crude product which was purified by silica gel column chromatography or prep-HPLC to afford the desired compound.
Method 2 (Using epoxide)
To a stirred solution of Het-Ar-OH compound (1 eq.) and corresponding epoxide compound (1.5/2 eq.) in DMF/ACN (6 mL/mmol) was added CS2CO3 (2.5 eq.). The reaction mixture was stirred at room temperature/ 60 °C for 4-12 h. The reaction progress was monitored by TLC. After completion, reaction mixture was diluted with water and extracted with ethyl acetate. The combined organic layers were collected, dried over anhydrous sodium sulphate, and concentrated under reduced pressure to obtain crude product which was purified by silica gel column chromatography or prep-HPLC to afford the desired compound.
Method for Suzuki coupling
Method 3
To a mixture of halo compound (1 eq.) and corresponding boronic acid/boronate ester (1.2- 1.5 eq.) in 1, 4-dioxane: water (4:1) (2.17 mL/mmol), Na2COs (2-3 eq.) was added and purged with Argon for 15 min. To this solution, Pd(dppf)Ch (0.1 eq.) was added and purged with Argon for another 10 min. The resulting reaction mixture was stirred at 100 °C for 12-16 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was filtered through Celite and evaporated to dryness. The residue was taken in ethyl acetate, washed with water, followed by brine, dried over anhydrous sodium sulphate, and evaporated under reduced pressure. The crude product was purified by either Combiflash column chromatography or prep-HPLC to afford the desired compound.
Method 4
To a mixture of halo compound (1 eq.) and corresponding boronic acid/boronate ester (1.2- 1.5 eq.) in 1, 4-dioxane: water (4: 1) (2.17 mL/mmol), K3PO4 (2-3 eq.) was added and purged with Argon for 15 min. To this solution, Pd(dppf)Ch (0.1 eq.) was added and purged with Argon for another 10 min. The resulting reaction mixture was stirred at 100 °C for 10 h-16 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was filtered through Celite and evaporated to dryness. The residue was taken in ethyl acetate, washed with water, followed by brine, dried over anhydrous sodium sulphate, and evaporated under reduced pressure. The crude product was purified by either Combiflash column chromatography or prep-HPLC to afford the desired compound.
Method 5
To a mixture of halo compound (1 eq.) and corresponding boronic acid/boronate ester (1.2- 1.5 eq.) in 1, 4-dioxane: water (4: 1) (2.17 mL/mmol), K3PO4 (2-3 eq.) was added and purged with Argon for 15 min. To this solution, S-Phos (0.2 eq.) and Pd(dppf)Ch (0.1 eq.) was added and purged with Argon for another 10 min. The resulting reaction mixture was stirred at 100 °C for 10 h-16 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was filtered through Celite and evaporated to dryness. The residue was taken in ethyl acetate, washed with water, followed by brine, dried over anhydrous sodium sulphate, and evaporated under reduced pressure. The crude product was purified by either Combiflash column chromatography or prep-HPLC to afford the desired compound.
Procedure for Sonogashira coupling
Method 6
To a mixture of halo compound (1 eq.) and corresponding alkyne compound (3 eq.) in dry THF (3 mL/mmol), Cui (0.2 eq.) and triethylamine (3 eq.) were added and purged with Argon for 15 min. To this solution, Pd(PPh3)2Ch (0.1 eq.) was added and purged with Argon for another 10 min. The resulting reaction mixture was stirred at room temperature for 12h-16h. The progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was filtered through Celite and evaporated to dryness. The residue was taken in ethyl acetate, washed with water, followed by brine, dried over anhydrous sodium sulphate, and evaporated under reduced pressure. The crude product was purified by either CombiFlash column chromatography or prep-HPLC to afford the desired compound.
Method 7
To a mixture of halo compound (1 eq.) in DMA: H2O (3:1), Cui (0.15 eq.), triethylamine (5 eq.) and K2CO3 (1.5 eq.) were added and purged with Argon for 15 min. To this solution, Pd(PPh3)4 (0.1 eq.) and corresponding alkyne compound (10 eq.) was added and purged with Argon for another 10 min. The resulting reaction mixture was stirred in microwave at 120 °C for 4h. The progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was filtered through Celite and evaporated to dryness. The crude product was purified by either CombiFlash column chromatography or prep-HPLC to afford the desired compound.
Procedure for Copper-Catalyzed N-Arylation
Method 8
To a mixture of halo compound (1 eq.) and corresponding amine (2.5 eq.) in DMSO (2 mL), K2CO3 (2.5 eq.) and L-proline (0.4 eq.) were added and purged with Argon for 10 min. To this solution, Cui (0.2 eq.) was added and purged with Argon for another 10 min. The resulting reaction mixture was stirred at 100°C for 16h. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was diluted with ice cold water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by CombiFlash column chromatography or prep-HPLC to afford the desired compound.
Procedure for reduction
Method 9
To a stirred solution nitro compound (1 eq.) in MeOH (20 mL/ mmol) at 0°C, 10% Pd/C and NaBH4 (2.5 eq.) were added. The reaction mixture was stirred at room temperature for 6 h. The progress of the reaction was monitored by TLC and LCMS. After completion, the reaction mixture was filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure. The crude product was purified by either silica gel column chromatography or prep-HPLC to afford the desired compound.
Procedure for amidation reaction
Method 10
To a stirred solution of acid compound (1.1-1.2 eq.) in DMF/dichloromethane (1.01 mL/mmol) at 0 °C, DIPEA (2-3 eq.) and HATU (1.5-2.5 eq.) were added and stirred for 5 min. To this solution, corresponding amine (1 eq.) was added. The resulting reaction mixture was stirred at room temperature for 12-16h. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was diluted with ice cold water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford a crude product. The crude product
was purified by either prep-HPLC or Combiflash column chromatography to afford the desired compound.
Method 11
To a stirred solution of acid compound (1.1-1.2 eq.) in DMF/Acetonitrile (1.01 mL/mmol) at 0 °C, Pyridine (5-10 eq.) and HATU (1.5-2.5 eq.) were added and stirred for 5 min. To this solution, corresponding amine (1-1.1.5 eq.) was added. The resulting reaction mixture was stirred at 80 °C for 12-16h. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was diluted with ice cold water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford a crude product. The crude product was purified by either prep-HPLC or Combiflash column chromatography to afford the desired compound.
Method 12 (amide coupling using acid chloride/derivatives)
To a stirred solution of amine compound (1 eq.) in dichloromethane/DMF (1.01 mL/mmol) was added triethylamine/DIPEA (1.5-3 eq.) at 0 °C and stirred for 5 min. To this solution, corresponding acid chloride/carbamic chloride/chloroformate (1.1-1.5 eq.) was added slowly at 0 °C and the reaction mixture was allowed to stir at room temperature till completion. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was diluted with ice cold water and extracted with ethyl acetate/dichloromethane. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford a crude product. The crude product was purified by either prep-HPLC or Combiflash column chromatography to afford the desired compound.
Method 13
To a stirred solution of ester compound (1 eq.) in methanol (1 mL/mmol), MeNH2 (2M in THF) was added. The reaction mixture was stirred at 70°C for 16 h. The progress of the reaction was monitored by TLC. After completion, the reaction mixture concentrated under reduced pressure to afford a crude product. The crude product was purified by silica gel column chromatography to afford the desired compound.
Procedure for \-acylatiun, V-Sulfonvlation, V-Sulfamovlation and V-Carbamovlation.
Method 14
To a stirred solution of Amine compound (1 eq.) in DMF (10 vol.) was added triethylamine or pyridine (5 eq.) followed by dropwise addition of corresponding acid chloride/ sulfonylchloride/ sulfamoyl chloride I carbamoylchloride (2.5 eq) at 0 °C. The reaction mixture was stirred at room temperature for 5-6 h. The progress of the reaction was monitored by TLC and LCMS. After completion, the reaction mixture was quenched with ice cold water and extracted with dichloromethane. The organic layer was concentrated under reduced pressure. The crude product was purified by either silica gel column chromatography or prep-HPLC to afford the desired compound.
Procedure for /V-alkylation
Method 15
To a stirred solution of amine compound (1 eq.) in THF (10 vol.) was added DIPEA (5 eq.) corresponding alkyl O-Tril’lale (2 eq) at 0 °C. The reaction mixture was stirred at room temperature for 30 min. The progress of the reaction was monitored by TLC and LCMS. After completion, the reaction mixture was quenched with ice cold water and extracted with dichloromethane. The organic layer was concentrated under reduced pressure. The crude product was purified by either silica gel column chromatography or prep-HPLC to afford the desired compound.
Procedure for Grignard Reaction
Method 16
To a stirred solution of keto compound (1 eq.) in dry THF (0.2 mL/mmol) in an inert atmosphere was added Grignard reagent (5 eq.) slowly via glass syringe at 0°C and stirred the reaction mixture at room temperature for 3h. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was diluted with sat. aq. solution of ammonium chloride and extracted with ethyl acetate/dichloromethane. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford a crude product. The crude product was purified by either by Combiflash column chromatography or prep-HPLC to afford the desired compound.
Procedure for Reductive amination
Method 17
To a stirred solution of aldehyde/keto compound (2 eq) in MeOH (0.1 mL/mmol) at 0 °C, AcOH (5 eq) was added and stirred at RT for 30 min. To this solution, amine (1 eq) and sodium cyano borohydride (5 eq) were added. The resulting reaction mixture was stirred at RT for overnight. The reaction progress was monitored by TLC. After completion reaction was diluted with sat. NaHCO solution and extracted with 10% MeOH/dichloromethane. The organic layer was separated, dried over anhydrous sodium sulphate, and concentrated in vacuo to obtain crude product. The crude product was purified by column chromatography to afford desired compound.
Intermediate 1

5-Oxo-l,3a,4,5,6,6a-hexahydropentalen-2-yl trifluoromethanesulfonate. To a solution of l,3,3a,4,6,6a-hexahydropentalene-2,5-dione (40.0g, 289.5 mmol) and pyridine (24.0 g, 304.0 mmol) in dichloromethane (600 ml) was added Tf2O (89.8 g, 318.5 mmol) dropwise at room temperature. The mixture was stirred at room temperature for 3 h. Brine (300 mL) was added, and the aqueous layer extracted with dichloromethane (200 mL x 3). The organic layer was separated, dried over Na2SC>4 and concentrated to give the crude product which was purified by silica gel column chromatography using 8:1 (v/v) petroleum ether/ethyl acetate to afford 5- oxo-l,3a,4,5,6,6a-hexahydropentalen-2-yl trifluoromethanesulfonate as a yellow oil. !H NMR (400 MHz, CDCh): 5 5.63 (q, J= 1.92 Hz, 1 H), 3.57 - 3.50 (m, 1 H), 3.14 - 3.00 (m, 2 H), 2.67 - 2.58 (m, 1 H), 2.56 -2.40 (m, 2 H), 2.34 - 2.26 (m, 1 H), 2.17 (ddd, 7 = 19.14, 7.34, 1.63 Hz, 1 H) ppm.
Intermediate 2
 5-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-3,3a,6,6a-tetrahydropentalen-2(lH)- one. A mixture of 5-oxo-l,3a,4,5,6,6a-hexahydropentalen-2-yl trifluoromethanesulfonate (110.0 g, 407.0 mmol), 4,4,5,5-tetramethyl-2-(4, 4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)- 1,3,2-dioxaborolane (108.5 g, 427.4 mmol), Pd(dppf)Ch (8.9 g, 12.2 mmol) and potassium acetate (119.7 g, 1221.0 mmol) in dioxane (1000 ml) was stirred at 80 °C under an N2 atmosphere for 2 h. The reaction mixture was filtered through a pad of Celite®545 and the filter cake was washed with EtOAc (250 mL x 3). The filtrate was concentrated under vacuo and the residue was purified by silica gel column chromatography using 8:1 petroleum ether/ethyl acetate to afford 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-3,3a,6,6a- tetrahydropentalen-2(lH)-one as a yellow oil. !H NMR (400 MHz, CDCI3): 5 6.37 (q, J = 2.08 Hz, 1 H), 3.54 - 3.41 (m, 1 H), 3.05 - 2.93 (m, 1 H), 2.79 (ddt, J= 16.48, 7.58, 2.64, 2.64 Hz, 1 H), 2.55 - 2.24 (m, 4 H), 2.07 - 1.95 (m, 1 H), 1.28 (s, 13 H) ppm.
5-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-3,3a,6,6a-tetrahydropentalen-2(lH)- one. A mixture of 5-oxo-l,3a,4,5,6,6a-hexahydropentalen-2-yl trifluoromethanesulfonate (110.0 g, 407.0 mmol), 4,4,5,5-tetramethyl-2-(4, 4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)- 1,3,2-dioxaborolane (108.5 g, 427.4 mmol), Pd(dppf)Ch (8.9 g, 12.2 mmol) and potassium acetate (119.7 g, 1221.0 mmol) in dioxane (1000 ml) was stirred at 80 °C under an N2 atmosphere for 2 h. The reaction mixture was filtered through a pad of Celite®545 and the filter cake was washed with EtOAc (250 mL x 3). The filtrate was concentrated under vacuo and the residue was purified by silica gel column chromatography using 8:1 petroleum ether/ethyl acetate to afford 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-3,3a,6,6a- tetrahydropentalen-2(lH)-one as a yellow oil. !H NMR (400 MHz, CDCI3): 5 6.37 (q, J = 2.08 Hz, 1 H), 3.54 - 3.41 (m, 1 H), 3.05 - 2.93 (m, 1 H), 2.79 (ddt, J= 16.48, 7.58, 2.64, 2.64 Hz, 1 H), 2.55 - 2.24 (m, 4 H), 2.07 - 1.95 (m, 1 H), 1.28 (s, 13 H) ppm.
Intermediate 3

Methyl 2,4-dibromo-l-methyl-lH-imidazole-5-carboxylate. To a solution of methyl l-methyl-lH-imidazole-5-carboxylate (16.6 g, 118.5 mmol) in CHCh (200 mL) was added NBS (78.3 g, 414.8 mmol) and AIBN (1.95 g, 11.9 mmol). The reaction mixture was stirred at 60 °C for 24 h. The mixture was concentrated and purified by column chromatography (Rf=0.4, petroleum ether: ethyl acetate=5:l) to give methyl 2,4-dibromo-l -methyl- 1H- imidazole-5-carboxylate (22.2 g, 63% yield) as a yellow solid.
Intermediate 4

N-(3-Chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide. To a solution of 1 -methyl- lH-imidazole-5 -carboxylic acid (10 g, 83 mmol), 3-chloro-4- fluoroaniline (18 g, 124 mmol) and EI3N (16 g, 160 mmol) in DMF (100 mL) was added
HATU (63 g, 160 mmol) at room temperature. The reaction mixture was stirred at 25 °C overnight then poured into water (200 mL). Yellow solids were formed from the solution which was filtered and dried to provide N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole- 5-carboxamide as a pale white solid. TLC; (50% ethyl acetate/petroleum ether) (Rf: 0.3). MS calcd. for C11H9CIFN3O: 253.0; Found: 254.1 [M+ 1]+.
Intermediate 5

2,4-Dibromo-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide (4 g, 15 mmol) in CHCI3 (100 mL) was added NBS (10 g, 60 mmol) and AIBN (0.25 g, 1.5 mmol) at room temperature. The reaction mixture was stirred at 50 °C for 18 h. The mixture was evaporated under vacuo to give a yellow residue. The residue was purified by silica gel chromatography to give 2,4-dibromo-N-(3-chloro-4-fluorophenyl)-l-methyl-lH- imidazole-5-carboxamide as yellow solid. TLC; 40% ethyl acetate I petroleum ether (Rf. 0.3). MS calcd. for CnH7Br2ClFN3O: 408.9; Found; 411.2 [M + 2]+.
Alternative synthesis of 2,4-Dibromo-N-(3-chloro-4-fluorophenyl)-l-methyl-lH- imidazole-5-carboxamide. To a solution of 2,4-dibromo-l-methyl-lH-imidazole-5- carboxylic acid (9.94 g, 35.0 mmol) in DMF (50 mL) was added HATU (13.3 g, 35.0 mmol) and DIPEA (9.69 g, 175 mmol) at 0 °C, the reaction mixture was stirred at 0 °C for 1 h. Then 3-chloro-4-fluoroaniline (6.1 g, 42.0 mmol) was added and the reaction mixture stirred at room temperature overnight. The mixture was added dropwise to water (600 mL), and the resulting precipitate filtered to provide 2,4-dibromo-N-(3-chloro-4-fluorophenyl)-l-methyl- lH-imidazole-5 -carboxamide (12.5 g, 87% yield) as a yellow solid.
Intermediate 6

4-Bromo-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide. To a solution of 2,4-dibromo-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide (1.1 g, 2.0 mmol) in THF (50 mL) was added CH Mgl (2 mL, 4.0 mmol) slowly at room temperature. The reaction mixture was stirred at 50 °C for 4 h then poured into water (50 ml) and extracted with ethyl acetate (20 mL x 3). The organic layer was dried and concentrated. The residue was purified by silica gel chromatography to give 4-bromo-N-(3- chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide as a yellow solid. TLC; 50% ethyl acetate/petroleum ether (Rf: 0.3). MS calcd. for Ci i HsBrCIFN O: 331.0; Found: 332.1 [M+ 1]+.
Alternative procedure for the synthesis of 4-bromo-N-(3-chloro-4-fluorophenyl)- l-methyl-lH-imidazole-5-carboxamide. The titled compound was synthesized following the general procedure described above for amidation (Method C) to afford 4-bromo-N-(3- chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide as a brown solid. TLC; 30% EtOAc/ hexanes (7? 0.45);
 NMR (DMSO-tfc, 400 MHz): 5 10.41 (s, 1H), 7.96 (dd, J = 6.8, 2.4 Hz, 1H), 7.85 (s, 1H), 7.63-7.60 (m, 1H), 7.43 (t, 7 = 9.6 Hz, 1H), 3.75 (s, 3H); MS calcd. for CnHsBrClFN O: 331.0; Found: 332.1 [M + 1]+.
NMR (DMSO-tfc, 400 MHz): 5 10.41 (s, 1H), 7.96 (dd, J = 6.8, 2.4 Hz, 1H), 7.85 (s, 1H), 7.63-7.60 (m, 1H), 7.43 (t, 7 = 9.6 Hz, 1H), 3.75 (s, 3H); MS calcd. for CnHsBrClFN O: 331.0; Found: 332.1 [M + 1]+.

N-(3-Chloro-4-fluorophenyl)-l-methyl-4-(5-oxo-l,3a,4,5,6,6a- hexahydropentalen-2-yl)-lH-imidazole-5-carboxamide. A mixture of 4-bromo-N-(3- chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide (600 mg, 1.8 mmol), 5-
(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-3,3a,6,6a-tetrahydropentalen-2(lH)-one (448 mg, 1.8 mmol), Pd(dppf)C12 (62 mg, 0.077 mmol) and K3PO4 (814 mg, 3.6 mmol) in dioxane (20 mL) and water (4 mL) was stirred at 100 °C for 4 h under N2. EtOAc (20 mL) was then added to the mixture. The mixture was filtered, and the filtrate washed with H2O (35 mL x 3). The organic layer was separated, dried over Na2SO4, and evaporated in vacuo to give a yellow residue. The residue was purified by silica gel column chromatography using 20 - 50% petroleum ether/ethyl acetate to give N-(3-chloro-4-fluorophenyl)-l-methyl-4-(5-oxo- l,3a,4,5,6,6a-hexahydropentalen-2-yl)-lH-imidazole-5-carboxamide as a brown solid. TLC; 5% MeOH/dichloromethane Rf. 0.2). MS calcd. for C19H17CIFN3O2: 373.13. Found; 374.1 [M+ 1]+.
Alternative synthesis of N-(3-Chloro-4-fluorophenyl)-l-methyl-4-(5-oxo- l,3a,4,5,6,6a-hexahydropentalen-2-yl)-lH-imidazole-5-carboxamide. To a solution of 4- bromo-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide (13.3 g, 40.0 mmol) in l,4-dioxane/H2O (v/v = 7/1 (v/v), 120 mL) were added 5-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-3,3a,6,6a-tetrahydropentalen-2(lH)-one (12.2 g, 48.0 mmol), Pd(dppf)C12 (2.9 g, 4.0 mmol) and Na2CC>3 (10.6 g, 100.0 mmol), respectively, and the mixture was stirred at 100 °C overnight. The reaction mixture was cooled to room temperature, filtered through a pad of Celite. The solid was washed with ethyl acetate and the filtrate was concentrated to give the crude product, which was purified by column chromatography on silica gel with 5% of methanol in dichloromethane (120 g silica gel column, 60 mL/min) to afford N-(3-chloro-4- fhiorophenyl)-l-methyl-4-(5-oxo- 1,3a, 4,5,6, 6a-hexahydropentalen-2-yl)-lH-imidazole-5- carboxamide (12.8 g, 85.6%) as a brown solid. TLC: 7% methanol/dichloromethane (Rf. 0.5); MS calcd. for C19H17CIFN3O2: 373.1; Found: 374.3 [M+ 1]+.
Example 1

N-(3-Chloro-4-fluorophenyl)-l-methyl-4-(5-oxooctahydropentalen-2-yl)-lH- imidazole-5 -carboxamide. To a solution of N-(3-chloro-4-fluorophenyl)-l-methyl-4-(5-oxo-
l,3a,4,5,6,6a-hexahydropentalen-2-yl)-lH-imidazole-5-carboxamide (300 mg, 0.8 mmol) in THF (20 ml) was added Pd/C (30 mg, 10% Pd). The mixture was stirred at 30 °C for 5 h under H2. The mixture was filtered, and the filtrate was evaporated in vacuo to give a yellow residue. The residue was purified by silica gel chromatography to give N-(3-chloro-4- fluorophenyl)- l-methyl-4-(5-oxooctahydropentalen-2-yl)- lH-imidazole-5-carboxamide a brown solid, as a single diastereomer. TLC; 50% ethyl acetate / petroleum ether (Rr. 0.3). MS calcd. for C19H19CIFN3O2: 375.2; Found; 376.2 [M + 1]+.
Alternative synthesis of N-(3-Chloro-4-fluorophenyl)-l-methyl-4-(5- oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide
To a solution of N-(3-chloro-4-fluorophenyl)-l-methyl-4-(5-oxo-l,3a,4,5,6,6a- hexahydropentalen-2-yl)-lH-imidazole-5-carboxamide (12.8 g, 34.2 mmol) in THF (200 mL) was added Pd/C (6.4 g, 10%) under H2 and the mixture stirred at room temperature for 4 hours. The mixture was filtered through a pad of Celite and washed with methanol. The filtrate was concentrated in vacuo to give the crude product, which was purified by column chromatography on silica gel with 5% of methanol in dichloromethane (80 g silica gel column, 50 mL/min) to afford N-(3-Chloro-4-fluorophenyl)-l-methyl-4-(5- oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide, a gray solid, as a single diastereomer. TLC: 7% methanol/dichloromethane (R/ 0.5); MS calcd. for C19H19CIFN3O2: 375.2; MS Found: 376.3 [M+ 1]+.
Intermediate 8
 l-Methyl-3-nitro-lH-pyrazole. NaOtBu (19.11 g, 199.1 mmol) was added to a stirred solution of 3-nitro-lH-pyrazole (15 g, 132.7 mmol) in DMF (150 mL) at 0 °C, and the reaction was stirred for 20 minutes. To this solution was added Mel (9.91 mL 159.24 mmol) drop wise. The resulting reaction mixture was stirred at RT for 16 h. After completion, the mixture was quenched with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography to
afford the title compound as an off-white solid. TLC: 20% EtOAc/hexane (R 0.2). !H NMR (400 MHz, DMSO-76): 57.98 (s, 1H), 7.03 (d, 7 = 2.0 Hz, 1H), 3.97 (s, 3H).
l-Methyl-3-nitro-lH-pyrazole. NaOtBu (19.11 g, 199.1 mmol) was added to a stirred solution of 3-nitro-lH-pyrazole (15 g, 132.7 mmol) in DMF (150 mL) at 0 °C, and the reaction was stirred for 20 minutes. To this solution was added Mel (9.91 mL 159.24 mmol) drop wise. The resulting reaction mixture was stirred at RT for 16 h. After completion, the mixture was quenched with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography to
afford the title compound as an off-white solid. TLC: 20% EtOAc/hexane (R 0.2). !H NMR (400 MHz, DMSO-76): 57.98 (s, 1H), 7.03 (d, 7 = 2.0 Hz, 1H), 3.97 (s, 3H).

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-nitro-lH-pyrazol-5- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. Under an inert atmosphere, LDA (2M in THF, 60 mL, 120 mmol) was added drop wise to a stirred solution of l-methyl-3-nitro-lH-pyrazole (10.16 g, 80 mmol) in dry THF (100 mL) at -78 °C, and the stirring continued for 2 h. To this was added a solution of N-(3-chloro-4-fluorophenyl)-l- methyl-4-(5-oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (3 g, 8 mmol) in THF at -78 °C. The mixture was stirred at -78 °C for 1 h. The reaction was then quenched with saturated NH4CI and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography to afford the title compound as an off-white solid. TLC: 5% MeOH/dichloromethane (R 0.3). MS calcd. for C23H24CIFN6O4: 502.15; Found: 503.3 [M+l]+.
Intermediate 10

4-(5-(3-Amino-l-methyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N- (3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide. Under a nitrogen atmosphere 10% Pd/C (0.5 g) and NaBIU (1.06 g, 27.88 mmol) were added to a stirred solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-nitro-lH-pyrazol-5- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (2 g, 3.98 mmol) in MeOH (20 mL). The reaction mixture was stirred at 0°C for 30 minutes. The reaction mixture was filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography to afford the title compound as an off-white solid. TLC: 10% MeOH/dichloromethane (R 0.1). MS calcd for C23H26CIFN6O2; 472.18. Found; 471.20 [M- 1]-. !H NMR (400 MHz, DMSO-<76): 5 10.25 (s, 1H), 7.95 (d, 7 = 4.4 Hz, 1H), 7.76 (s, 1H), 7.58-7.54 (m, 1H), 7.41 (t, 7 = 9.2 Hz, 1H), 6.62-5.57 (br.s, 2H), 5.39 (s, 1H), 5.17 (s, 1H), 3.69 (s, 6H), 3.32-3.31 (m, 1H, merged), 2.50-2.32(m, 2H, merged), 2.29-2.11 (m, 4H), 1.85- 1.83 (m, 4H).

Methyl 3-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4- yl)-2-hydroxyoctahydropentalen-2-yl)propiolate. In an inert atmosphere n-BiiLi (1.19 g, 18.6 mmol) was added to a stirred solution of methyl propionate (1.56 g, 18.6 mmol) in dry THF (40 mL) at -78 °C and the reaction mixture was stirred for 30 minutes. A solution of N- (3-chloro-4-fluorophenyl)-l-methyl-4-(5-oxooctahydropentalen-2-yl)-lH-imidazole-5- carboxamide (1 g, 2.66 mmol) in THF was added at -78 °C. The mixture was stirred at -78 °C for 2h. The reaction mixture was quenched with saturated NH4CI and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound as an off-white solid. TLC: 5% , J
 N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3-hydroxy-l-methyl-lH-pyrazol-5- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. Triethylamine (2 g, 19.82 mmol) and methyl 3-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH- imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)propiolate (1.3 g, 2.83 mmol) were added to a stirred solution of methyl hydrazine sulphate (2.85 g, 19.82 mmol) in EtOH (20 mL). The reaction mixture was stirred at 50 °C for 24 h. The mixture was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford the title compound as a white solid. TLC: 8%
N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3-hydroxy-l-methyl-lH-pyrazol-5- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. Triethylamine (2 g, 19.82 mmol) and methyl 3-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH- imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)propiolate (1.3 g, 2.83 mmol) were added to a stirred solution of methyl hydrazine sulphate (2.85 g, 19.82 mmol) in EtOH (20 mL). The reaction mixture was stirred at 50 °C for 24 h. The mixture was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford the title compound as a white solid. TLC: 8%

Ethyl 2-((5-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4- yl)-2-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-pyrazol-3-yl)oxy)propanoate. The title compound was synthesized from N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3- hydroxy-l-methyl-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide and ethyl 2-bromopropanoate according to method 1. TLC: 10% MeOH/dichloromethane (R 0.5). MS calcd. for C28H33CIFN5O5: 573.22; Found: 572.35 [M- 1]-.
Example 2

4-(5-(3-((l-Amino-l-oxopropan-2-yl)oxy)-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. A mixture of ethyl 2-((5-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l- methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-pyrazol-3- yl)oxy)propanoate (70 mg, 0.122 mmol) and methanolic ammonia (3 mL) was heated at 70°C for 12 h. The mixture was concentrated under reduced pressure. The crude product was purified by CombiFlash column chromatography followed by prep. HPLC to afford the title compound as an off-white solid. MS calcd. for C26H30CIFN6O4; 544.20. Found; 545.15 [M+l]+. !H NMR (400 MHz, DMSO-76): 5 10.22 (s, 1H), 7.98-7.94 (m, 1H), 7.65 (s, 1H), 7.56-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 7.26 (s, 1H), 7.10 (s, 1H), 5.53 (s, 1H), 5.25 (s, 1H), 4.67 (q, 7= 6.8 Hz, 1H), 3.72 (s, 3H), 3.67 (s, 3H), 3.25-3.20 (m, 1H), 2.50-2.44 (m, 2H), 2.15-2.06 (m, 4H), 1.88-1.80 (m, 4H), 1.34 (d, 7 = 6.4 Hz, 3H) ppm.
 4-(5-(3-(2-Amino-2-oxoethoxy)-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. Methanolic ammonia (2 mL) was added to a stirred solution of ethyl 2-((5-(5- (5-((3-chloro-4-fhiorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2- hydroxyoctahydropentalen-2-yl)-l -methyl- lH-pyrazol-3-yl)oxy)acetate (70 mg, 0.125 mmol) in THF (1 mL). The reaction mixture was stirred at 70°C for 10 h. The mixture was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound as an off-white solid. MS calcd. for C25H28CIFN6O4; 530.18. Found; 531.15 531.15 [M+l]+.
4-(5-(3-(2-Amino-2-oxoethoxy)-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. Methanolic ammonia (2 mL) was added to a stirred solution of ethyl 2-((5-(5- (5-((3-chloro-4-fhiorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2- hydroxyoctahydropentalen-2-yl)-l -methyl- lH-pyrazol-3-yl)oxy)acetate (70 mg, 0.125 mmol) in THF (1 mL). The reaction mixture was stirred at 70°C for 10 h. The mixture was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound as an off-white solid. MS calcd. for C25H28CIFN6O4; 530.18. Found; 531.15 531.15 [M+l]+.
 (400 MHz, DMSO-cfc): 5 10.20 (s, 1H), 7.96 (d, 7 = 4.8 Hz, 1H), 7.65 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 8.8 Hz, 1H), 7.30-7.22 (m, 2H), 5.56 (s, 1H), 5.26 (s, 1H), 4.38 (s, 2H), 3.73 (s, 3H), 3.67 (s, 3H), 3.27-3.21 (m, 1H), 2.50-2.42 (m, 2H), 2.19-2.06 (m, 4H), 1.90-1.80 (m, 4H) ppm.
(400 MHz, DMSO-cfc): 5 10.20 (s, 1H), 7.96 (d, 7 = 4.8 Hz, 1H), 7.65 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 8.8 Hz, 1H), 7.30-7.22 (m, 2H), 5.56 (s, 1H), 5.26 (s, 1H), 4.38 (s, 2H), 3.73 (s, 3H), 3.67 (s, 3H), 3.27-3.21 (m, 1H), 2.50-2.42 (m, 2H), 2.19-2.06 (m, 4H), 1.90-1.80 (m, 4H) ppm.

4-(5-(3-((l-Amino-3-methyl-l-oxobutan-2-yl)oxy)-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. The title compound was synthesized from N-(3-chloro-4-fluorophenyl)-4-(5- hydroxy-5-(3-hydroxy-l-methyl-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH- imidazole-5-carboxamide and 2-bromo-3-methylbutanamideaccording to Method 1. TLC: 7% MeOH/dichloromethane (R 0.4). MS calcd. for C28H34CIFN6O4; 572.23. Found; 555.20 [M- 18+l]+;. !H NMR (400 MHz, DMSO-tfo): 5 10.23 (s, 1H), 7.96 (dd, J = 6.8 Hz, 2.4 Hz, 1H), 7.65 (s. 1H), 7.58-7.54 (m, 1H), 7.40 (t, J = 9.2 Hz, 1H), 7.19 (s, 1H), 7.11 (s, 1H), 5.55 (s, 1H), 5.25 (s, 1H), 4.36 (d, J = 4.8 Hz, 1H), 3.71 (s, 3H), 3.67 (s, 3H), 3.28-3.22 (m, 1H), 2.48- 2.44 (m, 3H, merged), 2.18-2.06 (m, 4H), 1.90-1.80 (m, 4H), 0.96-0.90 (m, 6H) ppm.
Intermediate 14

4-(5-(3-Bromo-l-methyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N- (3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide. In an inert atmosphere n-BiiLi (2 M in THF, 7.8 mL, 15.96 mmol) was added dropwise to a stirred solution of 3,5- dibromo- l-melhyl- 1 //-pyrazole (3.8 g, 15.96 mmol) in dry THF (50 mL) at -78 °C and the mixture stirred for 35 minutes. To this was added a solution of N-(3-chloro-4-fluorophenyl)- l-methyl-4-(5-oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (1 g, 2.65 mmol) in THF slowly at -78°C. The mixture was allowed to regain room temperature and stirred for 16 h. The reaction mixture was diluted with sat. aq. ammonium chloride and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound. TLC: 5% MeOH/dichloromethane (R 0.3). !H NMR (400 MHz, DMSO-76): 5 10.21 (s, 1H), 7.95 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.65 (s, 1H), 7.58-7.55 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 6.23 (s, 1H), 5.37 (s, 1H), 3.87 (s, 3H), 3.67 (s, 3H), 3.29-3.23 (m, 1H), 2.50-2.46 (m, 2H, merged), 2.22-2.07 (m, 4H), 1.87- 1.83 (m, 4H) ppm. MS calcd. for C23H24BrClFN5O2; 535.08; Found: 536.10 [M+l]+.
Tables 1-5 show structures and analytical data for representative Examples of the invention. While the structures of the Examples shown throughout this specification are drawn without stereochemistry, unless otherwise specified they represent single isomer with stereochemistry consistent with the crystal structure shown below for reference compound AIA-227.
Table 1. Examples 5-8 were synthesized from 4-(5-(3-bromo-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide and the corresponding boronic acid according to Method 3, Method 4 or Method 5.



 N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methylpiperidin-3- yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer I. 20% Pd(OH)2 (20 mg) was added to a stirred solution of N-(3-chloro-4- fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methyl- 1,2,5, 6-tetr ahydropyridin-3-yl)- 1H- pyrazol-5-yl)octahydropentalen-2-yl)-l -methyl- lH-imidazole-5-carboxamide(55 mg, 0.099 mmol) in EtOAc (30 mL) in an autoclave, under nitrogen atmosphere. The reaction mixture was stirred at 40°C under a hydrogen atmosphere (100 psi) for 8 h. The reaction mixture was then filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure to the crude product was purified by prep-HPLC to afford the title compound (8 mg, 14.4%) as a mixture of enantiomers. TLC: 5% MeOH/dichloromethane (Rf. 0.4). The reaction was repeated on 150 mg scale to afford 80 mg of racemic compound which was purified for chiral prep HPLC purification to afford the pure enantiomers (Isomer I and Isomer II). N-(3-Chloro-4-fhiorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methylpiperidin-3- yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer
I and Isomer II. Isomer I MS calcd. for C29H36CIFN6O2; 554.26. Found; 553.60 [M-l]’. 1 H NMR (400 MHz, DMSO-tfo) 5 10.20 (s, 1H), 7.95 (d, 7 = 5.2 Hz, 1H), 7.65 (s, 1H), 7.60-7.52 (m, 1H), 7.40 (t, 7 = 9.6 Hz, 1H), 5.90 (s, 1H), 5.17 (s, 1H), 3.82 (s, 3H), 3.67 (s, 3H), 3.30- 3.20 (m, 2H), 2.87-2.82 (m, 1H), 2.72-2.60 (m, 2H), 2.45-2.40 (m, 3H), 2.10-2.05 (m, 7H), 1.90-1.72 (m, 6H), 1.65-1.50 (m, 2H) ppm.
N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methylpiperidin-3- yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer I. 20% Pd(OH)2 (20 mg) was added to a stirred solution of N-(3-chloro-4- fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methyl- 1,2,5, 6-tetr ahydropyridin-3-yl)- 1H- pyrazol-5-yl)octahydropentalen-2-yl)-l -methyl- lH-imidazole-5-carboxamide(55 mg, 0.099 mmol) in EtOAc (30 mL) in an autoclave, under nitrogen atmosphere. The reaction mixture was stirred at 40°C under a hydrogen atmosphere (100 psi) for 8 h. The reaction mixture was then filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure to the crude product was purified by prep-HPLC to afford the title compound (8 mg, 14.4%) as a mixture of enantiomers. TLC: 5% MeOH/dichloromethane (Rf. 0.4). The reaction was repeated on 150 mg scale to afford 80 mg of racemic compound which was purified for chiral prep HPLC purification to afford the pure enantiomers (Isomer I and Isomer II). N-(3-Chloro-4-fhiorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methylpiperidin-3- yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer
I and Isomer II. Isomer I MS calcd. for C29H36CIFN6O2; 554.26. Found; 553.60 [M-l]’. 1 H NMR (400 MHz, DMSO-tfo) 5 10.20 (s, 1H), 7.95 (d, 7 = 5.2 Hz, 1H), 7.65 (s, 1H), 7.60-7.52 (m, 1H), 7.40 (t, 7 = 9.6 Hz, 1H), 5.90 (s, 1H), 5.17 (s, 1H), 3.82 (s, 3H), 3.67 (s, 3H), 3.30- 3.20 (m, 2H), 2.87-2.82 (m, 1H), 2.72-2.60 (m, 2H), 2.45-2.40 (m, 3H), 2.10-2.05 (m, 7H), 1.90-1.72 (m, 6H), 1.65-1.50 (m, 2H) ppm.
 N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methylpiperidin-3- yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer II. MS calcd. for C29H36CIFN6O2; 554.26. Found; 555.70 [M+l]+.
N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methylpiperidin-3- yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer II. MS calcd. for C29H36CIFN6O2; 554.26. Found; 555.70 [M+l]+.
 NMR (400 MHz, DMSO-cfe) 5 10.20 (s, 1H), 7.96 (d, 7 = 4.4 Hz, 1H), 7.65 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 8.8 Hz, 1H), 5.91 (s, 1H), 5.18 (s, 1H), 3.82 (s, 3H), 3.67 (s, 3H), 2.92-2.88 (m, 1H), 2.80-2.74 (m, 1H), 2.70-2.64 (m, 1H), 2.49-2.42 (m, 2H), 2.20-2.06 (m, 8H), 1.94-1.80
NMR (400 MHz, DMSO-cfe) 5 10.20 (s, 1H), 7.96 (d, 7 = 4.4 Hz, 1H), 7.65 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 8.8 Hz, 1H), 5.91 (s, 1H), 5.18 (s, 1H), 3.82 (s, 3H), 3.67 (s, 3H), 2.92-2.88 (m, 1H), 2.80-2.74 (m, 1H), 2.70-2.64 (m, 1H), 2.49-2.42 (m, 2H), 2.20-2.06 (m, 8H), 1.94-1.80
(m, 8H), 1.64-1.50 (m, 2H) ppm.

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methyl-l, 2,3,6- tetrahydropyridin-4-yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH- imidazole-5-carboxamide. The above title was synthesized from 4-(5-(3-bromo-l-methyl- lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl- lH-imidazole-5 -carboxamide and (l-methyl-l,2,3,6-tetrahydropyridin-4-yl)boronic acid according to Method 4.

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methylpiperidin-4- yl)-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. In an autoclave, 10% Pd/C (20 mg) was added to a stirred solution of N-(3-chloro-4- fluorophenyl)-4-(5-hydroxy-5-(l-methyl-3-(l-methyl- 1,2,3, 6-tetrahydropyridin-4-yl)-lH-
pyrazol-5-yl)octahydropentalen-2-yl)-l -methyl- lH-imidazole-5-carboxamide (50 mg, 0.090 mmol) in EtOAc (10 mL) under nitrogen atmosphere. The reaction mixture was stirred in at 40°C under hydrogen atmosphere (100 psi) for 16 h. The reaction mixture was then filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure. The crude product was purified by prep-HPLC to afford the title compound as an off-white solid. MS calcd. for C29H36CIFN6O; 554.26. Found; 555.70 [M+l]+.
 (400 MHz, DMSO-cfe): 5 10.21 (s, 1H), 7.98-7.94 (m, 1H), 7.65 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 5.90 (s, 1H), 5.16 (s, 1H), 3.82 (s, 3H), 3.67 (s, 3H), 3.35-3.20 (m, 2H, merged), 2.88-2.82 (m, 2H), 2.45-2.36 (m, 2H), 2.23 (s, 3H), 2.20-2.15 (m, 2H), 2.08- 2.00 (m, 4H), 1.90-1.78 (m, 6H), 1.62-1.54 (m, 2H) ppm.
(400 MHz, DMSO-cfe): 5 10.21 (s, 1H), 7.98-7.94 (m, 1H), 7.65 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 5.90 (s, 1H), 5.16 (s, 1H), 3.82 (s, 3H), 3.67 (s, 3H), 3.35-3.20 (m, 2H, merged), 2.88-2.82 (m, 2H), 2.45-2.36 (m, 2H), 2.23 (s, 3H), 2.20-2.15 (m, 2H), 2.08- 2.00 (m, 4H), 1.90-1.78 (m, 6H), 1.62-1.54 (m, 2H) ppm.
Intermediate 16

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3-iodo-l-methyl-lH-pyrazol-5- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. LDA (2.8 g, 2.66 mmol) was added slowly to a stirred solution of 3-iodo- 1 -methyl- 1 H-pyrazole (5.5 g, 2.66 mmol) in dry THF (30 mL) at -78 °C and the mixture stirred for 1 h. To this solution was added N-(3-chloro-4-fluorophenyl)- l-methyl-4-(5-oxooctahydropentalen-2-yl)- 1H- imidazole-5-carboxamide (1 g, 2.66 mmol) at -78 °C. The resulting reaction mixture was stirred at RT for 2 h. The reaction was then quenched with saturated NH4CI solution and extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography using 1-2% MeOH in dichloromethane to afford the title compound as a white solid. TLC: 10% MeOH/dichloromethane (R 0.4). MS calcd. for C23H24CIFIN5O2; 583.06; Found; 584.05 [M+l]+; NMR (400 MHz, DMSO-76): 5 10.21 (s, 1H), 7.96 (d, 7 = 4.8 Hz, 1H), 7.65 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 8.0 Hz, 1H),
6.29 (s, 1H), 5.33 (s, 1H), 3.90 (s, 3H), 3.67 (s, 3H), 3.26-3.23 (m, 1H), 2.49-2.45 (m, 2H), 2.21-2.14 (m, 2H), 2.10-2.07 (m, 2H), 1.87-1.83 (m, 4H) ppm.

N -(3-Chlor o-4-fluorophenyl)-4-(5 -hydroxy-5 -(3-(3-hydroxy-3-methylbut- 1 -yn- 1 - yl)-l-methyl-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. The title compound was synthesized from N-(3-chloro-4-fluorophenyl)-4-(5- hydroxy-5-(3-iodo-l-methyl-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH- imidazole-5-carboxamide and 2-methylbut-3-yn-2-ol according to Method 6. TLC: 10% MeOH in dichloromethane (Rf. 0.5). MS calcd. for C28H31CIFN5O3; 539.21. Found; 522.15 [M-18+l]+. !H NMR (400 MHz, DMSO-76): 5 10.20 (s, 1H), 7.95 (d, 7 = 7.2 Hz, 1H), 7.70- 7.64 (m, 1H), 7.60-7.52 (m, 1H), 7.40 (t, 7 = 9.6 Hz, 1H), 6.20 (s, 1H), 5.40 (s, 1H), 5.33 (s, 1H), 3.88 (s, 3H), 3.67 (s, 3H), 3.28-3.20 (m, 1H), 2.48-2.40 (m, 2H), 2.24-2.16 (m, 2H),
2.12-2.02 (m, 2H), 1.90-1.76 (m, 4H), 1.41 (s, 6H) ppm.
Table 2. Examples 13-19 were synthesized from 4-(5-(3-bromo-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide and the corresponding boronic acid according to Method 6






Example 20

4-(5-(3-(6-(Aminomethyl)pyridin-3-yl)-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. Ammonium acetate (54.8 mg, 0.71 mmol) was added to a stirred solution of N- (3-chloro-4-fluorophenyl)-4-(5-(3-(6-formylpyridin-3-yl)-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (40 mg, 0.071 mmol) in MeOH (2 mL) at 0 °C and the mixture stirred at RT for 1 h. Sodium cyano borohydride (6.7 mg, 0.106 mmol) was then added and stirring continued at RT for 16 h. The reaction mixture was then concentrated under reduced pressure. The crude product was purified by prep. HPLC to afford the title compound as an off-white solid. TLC: 20% MeOH in dichloromethane (Rf. 0.1). MS calcd. for C29H31CIFN7O2: 563.22. Found: 564.03 [M+l] +. ' H NMR (400 MHz, CD3OD): 5 9.02 (s, 1H), 8.38-8.30 (m, 1H), 8.18 (d, 7 = 8.4 Hz, 1H), 7.92 (d, 7 = 4.4 Hz, 1H), 7.56-7.52 (m, 1H), 7.46 (d, 7= 8.0 Hz, 1H), 7.28 (t, 7= 8.8 Hz, 1H), 6.69 (s, 1H), 4.29 (s, 2H), 4.08 (s, 3H), 3.89 (s, 3H), 3.50-3.35 (m, 1H), 2.70-2.65 (m, 2H), 2.49- 2.35 (m, 4H), 2.11-1.90 (m, 4H) ppm. NH2 proton not observed.
Intermediate 18

4-Fluoro-3-iodo-l -methyl- 1/7-pyrazole. N-Iodosuccinimide (20.4 g, 90.69 mmol) was added to a stirred solution of 4-fluoro- 1 - 1 H-pyrazole (7.8 g, 90.69 mmol) in CHCI3 (120
mL). The reaction mixture was stirred at RT for 16 h. The reaction mixture was filtered, and the filtrate diluted with saturated sodium thiosulphate solution and extracted with chloroform. The organic layer was collected; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by CombiFlash column chromatography to afford 4-lluoro-3-iodo- 1 W-pyrazole as a white solid. TLC: 50%
 13.18 (s, 1H), 7.83 (s, 1H). To a stirred solution of 4-l’luoro-3-iodo- 1 W-pyrazole (6g, 28.3) in dry THF (60 mL at 0 °C) was added NaH (1.36 g, 33.9 mmol) in portions and the reaction mixture stirred at 0 °C for 5 min. Mel (8.81 mL, 141 mmol) was then added dropwise. The reaction mixture was stirred at RT for 2h. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound as an off-white solid. TLC: 20% EtOAc/Hexane (Rf: 0.6). !H NMR (400 MHz, DMSO-tfo): 57.82 (d, 7 = 8 Hz, 1H), 3.79 (s, 3H) ppm.
13.18 (s, 1H), 7.83 (s, 1H). To a stirred solution of 4-l’luoro-3-iodo- 1 W-pyrazole (6g, 28.3) in dry THF (60 mL at 0 °C) was added NaH (1.36 g, 33.9 mmol) in portions and the reaction mixture stirred at 0 °C for 5 min. Mel (8.81 mL, 141 mmol) was then added dropwise. The reaction mixture was stirred at RT for 2h. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound as an off-white solid. TLC: 20% EtOAc/Hexane (Rf: 0.6). !H NMR (400 MHz, DMSO-tfo): 57.82 (d, 7 = 8 Hz, 1H), 3.79 (s, 3H) ppm.

N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-iodo-l-methyl-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. LDA (5.33 mL, 10.66 mmol) was added dropwise to a stirred solution of 4-fluoro-3-iodo-l -methyl- 1H- pyrazole (2.42 g, 10.66 mmol) in dry THF (30 mL) at -78°C and the reaction mixture stirred for 2 h. To this, a solution of N-(3-chloro-4-fhiorophenyl)-l-methyl-4-(5- oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (0.4 g, 1.06 mmol) in THF was added at -78 °C. The reaction mixture was allowed to warm to room temperature and stirred for 3h. The reaction was then quenched with saturated NH4CI and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica
gel column chromatography to afford the title compound as an off-white solid. TLC: 5% MeOH/ dichloromethane (R 0.3). NMR (400 MHz, DMSO-76): 510.20 (s, 1H), 7.98-7.94 (m, 1H), 7.65 (s, 1H), 7.60-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 5.44 (s, 1H), 3.89 (s, 3H), 3.67 (s, 3H), 3.28-3.18 (m, 1H), 2.55-2.40 (m, 2H, merged), 2.24-2.18 (m, 2H), 2.10-2.06 (m, 2H), 1.98-1.90 (m, 4H) ppm. MS calcd. for C23H23CIF2IN5O2: 601.06; Found: 602.10
[M+l]+.
Table 3. Examples 21-23 were synthesized from N-(3-Chloro-4-fhiorophenyl)-4-(5-(4-fluoro-
3-iodo-l-methyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH- imidazole-5-carboxamide and the corresponding boronic acid according to Method 6



N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-((2-hydroxy-2- methylpropyl)amino)-l-methyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l- methyl-lH-imidazole-5-carboxamide. K2CO3 (99 mg, 0.415 mmol) and L-proline (7.6 mg, 0.066 mmol) were added to a mixture of N-(3-chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-iodo- l-methyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide (100 mg, 0.166 mmol) and l-amino-2-methylpropan-2-ol (29.6 mg, 0.33 mmol) in DMSO (3 mL) which was then purged with Argon for 10 min. To this solution, Cui (6.3 mg, 0.033 mmol) was added, and purging continued for another 10 min. The resulting reaction mixture was stirred at 90 °C for 16 h. The mixture was diluted with water and extracted with 10% MeOH/dichloromethane. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by prep. HPLC to afford the title compound. TLC: 10% MeOH/dichloromethane (R 0.2). MS calcd. for C27H33CIF2N6O3: 562.23. Found: 563.20 [M+l]+. !H NMR (400 MHz, DMSO-tfo): 5 10.21 (s, 1H), 7.95 (d, J = 6.8 Hz, 1H), 7.64 (s, 1H), 7.58-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 5.25 (s, 1H), 4.53 (t, 7 = 6.4 Hz, 1H), 4.45 (s, 1H), 3.67 (s, 3H), 3.64 (s, 3H), 3.26-3.22 (m, 1H), 2.94 (d, 7= 6.0 Hz, 2H), 2.55-2.40 (m, 2H, merged), 2.25-2.20 (m, 2H), 2.12-2.06 (m, 2H), 1.98-1.85 (m, 4H), 1.10 (s, 6H) ppm.
Intermediate 20

2-(3-Nitro-lH-pyrazol-l-yl)ethan-l-ol. To a stirred solution of compound 3-nitro- IH-pyrazole (5 g, 44.247 mmol) in THF (50 mL) was added, K2CO3 (12.22 g, 88.50 mmol) followed by 2-bromoethan-l-ol (8.22 g, 66.371 mmol) dropwise. The resulting reaction mixture was stirred at 70 °C for 16 h. After completion, the reaction mixture was diluted with water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh, using a gradient method of 80-90% EtOAc in hexane) to afford the title compound as an off white solid. TLC: 60% EtOAc/hexane (R 0.2).
Intermediate 21
 l-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-3-nitro-lH-pyrazole. To a stirred solution of compound 2-(3-nitro- 1 H-pyrazol- l-yl)ethan- l-ol (2 g, 12.740 mmol) in dichloromethane (20 mL) was added imidazole (1.30 g, 19.108 mmol) followed by TBDMS-C1 (2.30 g 15.286 mmol) at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred for 2 h. After completion the reaction mixture was diluted with water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh, using a gradient method of 40-50% EtOAc in hexane) to afford the title compound as an off white solid. TLC: 50% EtOAc/hexane (R 0.5).
Intermediate 22
l-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-3-nitro-lH-pyrazole. To a stirred solution of compound 2-(3-nitro- 1 H-pyrazol- l-yl)ethan- l-ol (2 g, 12.740 mmol) in dichloromethane (20 mL) was added imidazole (1.30 g, 19.108 mmol) followed by TBDMS-C1 (2.30 g 15.286 mmol) at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred for 2 h. After completion the reaction mixture was diluted with water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh, using a gradient method of 40-50% EtOAc in hexane) to afford the title compound as an off white solid. TLC: 50% EtOAc/hexane (R 0.5).
Intermediate 22

4-(5-(l-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-3-nitro-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. To a stirred solution of compound l-(2-((/er/-butyldimethylsilyl)oxy)ethyl)-3- nilro- 1 //-pyrazole (23.12 g, 85.33 mmol) in dry THF (50 mL) at -78 °C in an inert atmosphere, LDA (2M in THF, 53.33 mL, 106.66 mmol) was added dropwise and the reaction mixture stirred at -78 °C for 2 h. To this, a solution of N-(3-chloro-4-fluorophenyl)- l-methyl-4-(5-oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (4.0 g, 10.66 mmol) in THF was added at -78 °C and the resulting reaction mixture was stirred at -78 °C for 2 h. After completion, the reaction was quenched with saturated NH4CI and extracted with EtOAc. The combined organic layers were washed with brine; dried over anhydrous Na2SC>4, filtered, and concentrated under reduced pressure. The crude compound was purified by Combiflash® column chromatography (using a gradient method of 1-2% MeOH in DCM) to afford the title compound as a pale yellow solid. TLC: 10% MeOH/DCM (R 0.3). 1 H NMR (400 MHz, DMSO-<76): 5 10.21 (s, 1H), 8.01 - 7.91 (m, 1H), 7.70 - 7.62 (m, 1H), 7.61 - 7.52 (m, 1H), 7.45 - 7.34 (m, 1H), 6.95 - 6.88 (m, 1H), 5.73 - 5.66 (m, 1H), 4.55 - 4.44 (m, 2H), 4.11 - 4.00 (m, 2H), 3.68 (s, 3H), 2.45 - 2.37 (m, 2H), 2.17 - 2.05 (m, 2H), 1.89 - 1.68 (m, 3H), 1.65 - 1.53 (m, 1H), 1.34 - 1.09 (m, 2H), 0.97 - 0.64 (m, 16H); MS calcd. for C3oH4oClFN605Si: 646.25, Found: 647.55 [M+l]+.

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-(2-hydroxyethyl)-3-nitro-lH- pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide To a stirred solution of 4-(5-(l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-3-nitro-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide (0.55 g, 0.892 mmol) in THF (15 mL) under nitrogen atmosphere, was added TBAF (0.7 g, 2.678 mmol) at 0 °C and the reaction mixture was stirred at room temperature for 2 h. After completion, the reaction mixture was quenched with aq. NaHCO and extracted with dichloromethane. The combined organic layers were dried over anhydrous Na2SC>4, filtered, and concentrated under reduced pressure. The crude compound was purified by column chromatography on silica gel (100-200 mesh, using a gradient method of 1-2% MeOH in dichloromethane) to afford the title compound as an off white solid. TEC: 10% MeOH/dichloromethane (R 0.4).
Example 25
 4-(5-(3-Amino-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. To a stirred solution of 4-(5-(l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-3-nitro- lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl- lH-imidazole-5 -carboxamide (0.55g, 0.892 mmol) in THF (15 mL) under nitrogen atmosphere, was added TBAF (0.7g, 2.678 mmol) and the mixture was stirred at RT for 2h. The reaction mixture was then quenched with aq. NaHCO and extracted with dichloromethane. The organic layer was dried over Na2SC>4 and concentrated under reduced pressure to give a thick oil. The crude product was purified by silica gel column chromatography to afford alcohol compound (0.247g, 56%) as an off-white solid. TLC: 10% MeOH/dichloromethane (R 0.4). MS calcd. for C24H28CIFN6O3: 502.19. Found: 501.20 [M- 1]_. 10% Pd/C (15mg) and NaBFU (35mg, 0.92 mmol) were added to a stirred solution of the alcohol (0.1 g, 0.187 mmol) in MeOH (10 mL) under nitrogen atmosphere. The reaction mixture was stirred at RT for 5h. The reaction mixture was then filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography followed by prep-HPLC to afford the title compound as an off-white solid. TLC: 10% MeOH/dichloromethane (R 0.1).
4-(5-(3-Amino-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. To a stirred solution of 4-(5-(l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-3-nitro- lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl- lH-imidazole-5 -carboxamide (0.55g, 0.892 mmol) in THF (15 mL) under nitrogen atmosphere, was added TBAF (0.7g, 2.678 mmol) and the mixture was stirred at RT for 2h. The reaction mixture was then quenched with aq. NaHCO and extracted with dichloromethane. The organic layer was dried over Na2SC>4 and concentrated under reduced pressure to give a thick oil. The crude product was purified by silica gel column chromatography to afford alcohol compound (0.247g, 56%) as an off-white solid. TLC: 10% MeOH/dichloromethane (R 0.4). MS calcd. for C24H28CIFN6O3: 502.19. Found: 501.20 [M- 1]_. 10% Pd/C (15mg) and NaBFU (35mg, 0.92 mmol) were added to a stirred solution of the alcohol (0.1 g, 0.187 mmol) in MeOH (10 mL) under nitrogen atmosphere. The reaction mixture was stirred at RT for 5h. The reaction mixture was then filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography followed by prep-HPLC to afford the title compound as an off-white solid. TLC: 10% MeOH/dichloromethane (R 0.1).

4-(5-(3-Amino-4-chloro-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. N-Chlorosuccinimide (10 mg, 0.075 mmol) was added to a stirred solution of
4-(5-(3-amino-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3- chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide SL-AIA-1222 (25 mg, 0.0498 mmol) in DMF (1.5 mL). The reaction mixture was stirred at RT for 2 h. The reaction was quenched with ice cold water and extracted with 5% MeOH/dichloromethane. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by CombiFlash column chromatography followed by prep. HPLC to afford the title compound as a white solid. TLC: 5% MeOH/dichloromethane (R 0.3). MS calcd. for C24H27CI2FN6O3: 536.15. Found: 519.10 [M-18+l]+. !H NMR (400 MHz, DMSO-tfo): 5 10.22 (s, 1H), 7.96 (d, 7 = 5.6 Hz, 1H), 7.64 (s, 1H), 7.60-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 5.29 (s, 1H), 4.95-4.90 (m, 1H), 4.62 (s, 2H), 4.18 (t, 7 = 6.4 Hz, 2H), 3.70-3.60 (m, 5H), 3.30-3.15 (m, 1H), 2.60-2.55 (m, 2H), 2.42- 2.30 (m, 2H), 2.08-1.90 (m, 6H) ppm.
Intermediate 24

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-(2-methyl-2- ((triethylsilyl)oxy)propyl)-3-nitro-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl- lH-imidazole-5-carboxamide. LDA (1.99 g, 18.66 mmol) was added slowly to a stirred solution of l-(2-methyl-2-((triethylsilyl)oxy)propyl)-3-nitro-lH-pyrazole (5.58 g, 1.866 mmol) in dry THF (40 mL) at -78°C in an inert atmosphere, and the reaction mixture stirred for 2 h. To this, a solution of N-(3-chloro-4-fhiorophenyl)-l-methyl-4-(5- oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (0.7 g, 1.86 mmol) in THF was added at -78°C. The resulting reaction mixture was stirred at -78°C for 2 h. The reaction was quenched with saturated NH4CI and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to
afford the title compound. TLC: 10% MeOH/dichloromethane (R 0.3). MS Calculated for C32H44ClFN6O5Si: 674.28; Found: 675.05 [M+l]+.

4-(5-(3-Amino-l-(2-hydroxy-2-methylpropyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. 10% Pd/C (70 mg) and NaBPU (96 mg, 2.59 mmol) were added to a stirred solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-(2-methyl-2- ((triethylsilyl)oxy)propyl)-3-nitro-lH-pyrazol-5-yl)octahydropentalen-2-yl)-l-methyl-lH- imidazole-5-carboxamide (0.7 g, 1.04 mmol) in MeOH (20 mL) under nitrogen atmosphere. The mixture was stirred at RT for 6 h. The reaction mixture was then filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound. TLC: 10% MeOH/dichloromethane (R 0.2). MS calcd. for C26H32CIFN6O3: 530.22; Found: 529.45 [M-l]-.
 4-(5-(3-Amino-4-chloro-l-(2-hydroxy-2-methylpropyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. N-Chlorosuccinimide (45.34 mg, 0.339 mmol) was added to a stirred solution of 4-(5-(3-amino-l-(2-hydroxy-2-methylpropyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide (0.15 g, 0.283 mmol) in ACN (10 mL). The reaction mixture was stirred at RT for 12 h. The reaction was quenched with saturated NaHCOa and extracted with 10% MeOH/dichloromethane. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by prep. HPLC to afford the title compound as a white solid. TLC: 10% MeOH/dichloromethane (R 0.4). MS calcd. for C26H31CI2FN6O3: 564.18. Found: 565.20 [M+l]+. !H NMR (400 MHz, DMSO-tfo): 5 10.21 (s, 1H), 7.96 (dd, 7 = 6.4 Hz, 2.0 Hz, 1H), 7.64 (s, 1H), 7.60-7.55 (m, 1H), 7.41 (t, 7 = 9.6 Hz, 1H), 5.70 (s, 1H), 5.60 (s, 1H), 4.73 (s, 2H), 4.15 (s, 2H), 3.67 (s, 3H), 3.24-3.15 (m, 1H), 2.65-2.55 (m, 2H), 2.45-2.36 (m, 2H), 2.10-1.95 (m, 4H), 1.90-1.82 (m, 2H), 1.07 (s, 6H) ppm.
4-(5-(3-Amino-4-chloro-l-(2-hydroxy-2-methylpropyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. N-Chlorosuccinimide (45.34 mg, 0.339 mmol) was added to a stirred solution of 4-(5-(3-amino-l-(2-hydroxy-2-methylpropyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide (0.15 g, 0.283 mmol) in ACN (10 mL). The reaction mixture was stirred at RT for 12 h. The reaction was quenched with saturated NaHCOa and extracted with 10% MeOH/dichloromethane. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by prep. HPLC to afford the title compound as a white solid. TLC: 10% MeOH/dichloromethane (R 0.4). MS calcd. for C26H31CI2FN6O3: 564.18. Found: 565.20 [M+l]+. !H NMR (400 MHz, DMSO-tfo): 5 10.21 (s, 1H), 7.96 (dd, 7 = 6.4 Hz, 2.0 Hz, 1H), 7.64 (s, 1H), 7.60-7.55 (m, 1H), 7.41 (t, 7 = 9.6 Hz, 1H), 5.70 (s, 1H), 5.60 (s, 1H), 4.73 (s, 2H), 4.15 (s, 2H), 3.67 (s, 3H), 3.24-3.15 (m, 1H), 2.65-2.55 (m, 2H), 2.45-2.36 (m, 2H), 2.10-1.95 (m, 4H), 1.90-1.82 (m, 2H), 1.07 (s, 6H) ppm.
Intermediate 25
 l-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-4-f'luoro-3-iodo-lH-pyrazole The title compound has been synthesized from 4-fluoro-3-iodo-lH-pyrazoleand (2-bromoethoxy)(tert- butyl)dimethylsilane according Method 1. TLC: 30% EtOAc/hexane (Rf. 0.4); 1 H NMR (400 MHz, DMSO-tfo): 57.81 (d, 7 = 4.8 Hz, 1H), 4.11 (t, 7 = 4.8 Hz, 2H), 3.86 (t, 7 = 4.8 Hz, 2H), 0.78 (s, 9H), 0.08 (s, 6H) ppm.
Example 29
l-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-4-f'luoro-3-iodo-lH-pyrazole The title compound has been synthesized from 4-fluoro-3-iodo-lH-pyrazoleand (2-bromoethoxy)(tert- butyl)dimethylsilane according Method 1. TLC: 30% EtOAc/hexane (Rf. 0.4); 1 H NMR (400 MHz, DMSO-tfo): 57.81 (d, 7 = 4.8 Hz, 1H), 4.11 (t, 7 = 4.8 Hz, 2H), 3.86 (t, 7 = 4.8 Hz, 2H), 0.78 (s, 9H), 0.08 (s, 6H) ppm.
Example 29

N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-l-(2-hydroxyethyl)-3-iodo-lH- pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. LDA (2M in THF, 13.3 mL, 26.6 mmol) was added dropwise to a stirred solution of l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-4-fluoro-3-iodo-lH-pyrazole (7.89g, 21.32 mmol) in dry THF (150 mF) at -78 °C in an inert atmosphere and the reaction mixture was stirred for 2 h. To this, a solution of N-(3-chloro-4-fluorophenyl)-l-methyl-4-(5- oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (1 g, 2.66 mmol) in THF was added at -78 °C. The resulting reaction mixture was stirred at RT for 16 h. The reaction was then quenched with saturated NH4CI solution and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the title compound as an off-white solid. TLC: 5% MeOH/dichloromethane (Rf: 0.4). MS Calculated for C24H25CIF2IN5O3: 631.07; Found: 631.90 [M+l]+.

Example 31

N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-l-(2-hydroxyethyl)-3-(l, 2,5,6- tetrahydropyridin-3-yl)-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl- lH-imidazole-5-carboxamide. SnCh (11.3 mg, 0.047 mmol) was added to a stirred solution
of tert-butyl 5-(5-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)- 1-methyl- lH-imidazol-4-yl)-2- hydroxyoctahydropentalen-2-yl)-4-fluoro-l-(2-hydroxyethyl)-lH-pyrazol-3-yl)-3,6- dihydropyridine-l(2H)-carboxylate (30 mg, 0.043 mmol) in ACN (5 mL) at 0°C. The reaction mixture was stirred at 0°C for 30 min. The reaction mixture was diluted with sat. NaHCCh and extracted with ethyl acetate. The organic layer was washed with water, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified by prep. HPLC to afford the title compound as an off-white solid; TLC: 10% MeOH in dichloromethane (Rf. 0.1). LC MS calcd. for C29H33CIF2N6O3: 586.23. Found: 587.15 [M+l]+. !H NMR (400 MHz, DMSO-tfo): 5 10.22 (s, 1H), 7.96 (d, 7 = 5.2 Hz, 1H), 7.64 (s, 1H), 7.60-7.54 (m, 1H), 7.40 (t, 7 = 9.6 Hz, 1H), 6.25 (s, 1H), 5.43 (s, 1H), 4.92-4.88 (m, 1H), 4.24-4.22 (m, 2H), 3.75-3.60 (m, 7H), 3.50-3.20 (m, 1H, merged), 2.94-2.92 (m, 2H), .60-2.40 (m, 2H, merged), 2.35-2.20 (m, 4H), 2.15-2.08 (m, 2H), 1.98-1.92 (m, 4H) ppm. NH proton is not observed.

N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-(4-hydroxycyclohex-l-en-l-yl)-l- (2-hydroxyethyl)-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH- imidazole-5 -carboxamide. The title compound was synthesized from N-(3-chloro-4- fluorophenyl)-4-(5 -(4-fluoro- 1 -(2-hydroxyethyl)-3 -iodo- 1 H-pyrazol-5 -yl)-5 - hydroxyoctahydropentalen-2-yl)- 1-methyl- lH-imidazole-5-carboxamide and dimethyl cyclohex- 1-en-l-ylboronate according to Method 4. MS calcd for C30H34CIF2N5O4: 601.23. Found: 602.10 [M+l]+.

N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-(4-hydroxycyclohexyl)-l-(2- hydroxyethyl)-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH- imidazole-5 -carboxamide Isomer I. In an autoclave, 10% Pd/C (30 mg) was added to a stirred solution of N-(3-chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-(4-hydroxycyclohex-l-en-l- yl)-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH- imidazole-5-carboxamide (lOOmg, 0.16 mmol) in EtOAc (10 mL) under nitrogen atmosphere. The reaction mixture was stirred in at 45°C under hydrogen atmosphere (100 psi) for 36 h. The reaction mixture was filtered through a pad of Celite and washed with methanol. The filtrate was concentrated under reduced pressure. The crude product was purified by prep-HPLC to afford the compound N-(3-chloro-4-fluorophenyl)-4-(5-(4-fluoro- 3-(4-hydroxycyclohexyl)-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen- 2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer I and Isomer II. Isomer I; MS calcd. for C30H36CIF2N5O4: 603.24. Found: 586.20 [
 5 10.21 (s, 1H), 7.97 (dd, 7 = 6.8 Hz, 2.4 Hz, 1H), 7.64 (s, 1H), 7.58-7.55 (m, 1H), 7.41 (t, 7 = 8.8 Hz, 1H), 5.35 (s, 1H), 4.88 (t, 7 = 5.2 Hz, 1H), 4.53 (d, 7 = 4.4 Hz, 1H), 4.19 (t, 7 = 6.4 Hz, 2H), 3.72-3.67 (m, 5H), 3.39-3.21 (m, 3H), 2.55-2.40 (m, 2H, merged), 2.24-2.20 (m, 2H), 2.10-2.06 (m, 2H), 1.94-1.80 (m, 8H), 1.48-1.44 (m, 2H), 1.24-1.18 (m, 2H) ppm.
5 10.21 (s, 1H), 7.97 (dd, 7 = 6.8 Hz, 2.4 Hz, 1H), 7.64 (s, 1H), 7.58-7.55 (m, 1H), 7.41 (t, 7 = 8.8 Hz, 1H), 5.35 (s, 1H), 4.88 (t, 7 = 5.2 Hz, 1H), 4.53 (d, 7 = 4.4 Hz, 1H), 4.19 (t, 7 = 6.4 Hz, 2H), 3.72-3.67 (m, 5H), 3.39-3.21 (m, 3H), 2.55-2.40 (m, 2H, merged), 2.24-2.20 (m, 2H), 2.10-2.06 (m, 2H), 1.94-1.80 (m, 8H), 1.48-1.44 (m, 2H), 1.24-1.18 (m, 2H) ppm.

N-(3-Chloro-4-fhiorophenyl)-4-(5-(4-fhioro-3-(4-hydroxycyclohexyl)-l-(2- hydroxyethyl)-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH- imidazole-5 -carboxamide Isomer II. !H NMR (400 MHz, DMSO-7e): 5 10.21 (s, 1H), 7.97 (dd, 7 = 6.4 Hz, 2.4 Hz, 1H), 7.64 (s, 1H), 7.60-7.54 (m, 1H), 7.41 (t, 7 = 8.8 Hz, 1H), 5.35 (s, 1H), 4.90-4.85 (m,lH), 4.29 (d, 7 = 3.2 Hz, 1H), 4.20 (t, 7 = 6.0 Hz, 2H), 3.80-3.76 (m, 1H), 3.74-3.69 (m, 2H), 3.68 (s, 3H), 3.28-3.20 (m, 2H), 2.60-2.45 (m, 2H, merged), 2.30-2.25(m, 2H), 2.12-2.06 (m, 2H), 1.98-1.86 (m, 6H), 1.70-1.62 (m, 2H), 1.52-1.46 (m, 4H) ppm.
 N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-((2-hydroxy-2- methylpropyl)amino)-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. K2CO3 (32.7 mg, 0.237 mmol) and L-proline (3.63 mg, 0.031 mmol) were added to a mixture of N-(3-
chloro-4-fluorophenyl)-4-(5-(4-fluoro-l-(2-hydroxyethyl)-3-iodo-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (50 mg, 0.079 mmol) and l-amino-2-methylpropan-2-ol (14.1 mg, 0.158 mmol) in DMSO (2 mL) and the solution purged with Argon for 10 min. To this solution was added Cui (3 mg, 0.016 mmol) and purging with Argon continued for another 10 min. The resulting reaction mixture was stirred at 100°C for 16h. The reaction mixture was then diluted with ice cold water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by CombiFlash column chromatography to afford the title compound. TLC:5% MeOH/dichloromethane (R 0.3). MS calcd for C28H35CIF2N6O4: 592.24; Found: 593.05 [M+l]+. !H NMR (400 MHz, DMSO-tfo): 5 10.20 (s, 1H), 7.96 (d, 7 = 6.8 Hz, 1H), 7.65 (s, 1H), 7.60-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 5.31 (s, 1H), 4.84-4.80 (m, 1H), 4.61 (t, 7 = 4.8 Hz, 1H), 4.46 (s, 1H), 4.05 (t, 7 = 6.4 Hz, 2H), 3.70-3.62 (m, 5H), 3.25-3.22 (m, 1H), 2.96 (d, 7 = 6.4 Hz, 2H), 2.60-2.50(m, 2H, merged), 2.28-2.22 (m, 2H), 2.10-2.06 (m, 2H), 2.00- 1.88 (m, 4H), 1.10 (s, 6H) ppm.
N-(3-Chloro-4-fluorophenyl)-4-(5-(4-fluoro-3-((2-hydroxy-2- methylpropyl)amino)-l-(2-hydroxyethyl)-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. K2CO3 (32.7 mg, 0.237 mmol) and L-proline (3.63 mg, 0.031 mmol) were added to a mixture of N-(3-
chloro-4-fluorophenyl)-4-(5-(4-fluoro-l-(2-hydroxyethyl)-3-iodo-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (50 mg, 0.079 mmol) and l-amino-2-methylpropan-2-ol (14.1 mg, 0.158 mmol) in DMSO (2 mL) and the solution purged with Argon for 10 min. To this solution was added Cui (3 mg, 0.016 mmol) and purging with Argon continued for another 10 min. The resulting reaction mixture was stirred at 100°C for 16h. The reaction mixture was then diluted with ice cold water and extracted with ethyl acetate. The organic layer was collected; washed with brine; dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by CombiFlash column chromatography to afford the title compound. TLC:5% MeOH/dichloromethane (R 0.3). MS calcd for C28H35CIF2N6O4: 592.24; Found: 593.05 [M+l]+. !H NMR (400 MHz, DMSO-tfo): 5 10.20 (s, 1H), 7.96 (d, 7 = 6.8 Hz, 1H), 7.65 (s, 1H), 7.60-7.54 (m, 1H), 7.40 (t, 7 = 9.2 Hz, 1H), 5.31 (s, 1H), 4.84-4.80 (m, 1H), 4.61 (t, 7 = 4.8 Hz, 1H), 4.46 (s, 1H), 4.05 (t, 7 = 6.4 Hz, 2H), 3.70-3.62 (m, 5H), 3.25-3.22 (m, 1H), 2.96 (d, 7 = 6.4 Hz, 2H), 2.60-2.50(m, 2H, merged), 2.28-2.22 (m, 2H), 2.10-2.06 (m, 2H), 2.00- 1.88 (m, 4H), 1.10 (s, 6H) ppm.
Table 4. Examples 36 were synthesized according to the methods described elsewhere in this case.













































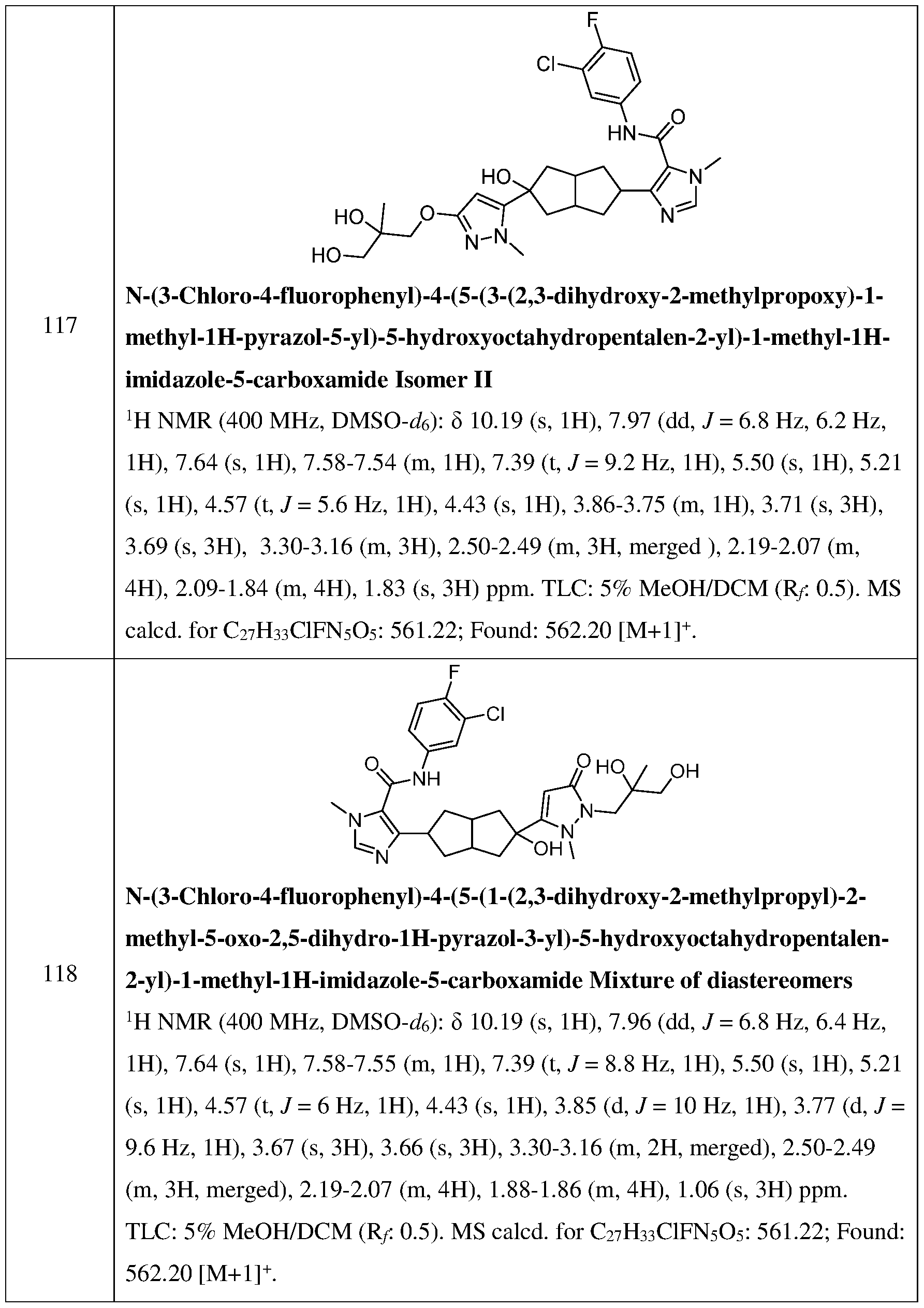


















N -(3-Chlor o-4-fluorophenyl)-4-(5 -hydroxy-5 -(1 -( l-methylpiperidin-4-yl)-3- (trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3- (trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide 200 mg, 0.39 mmol), CS2CO3 (140 mg, 0.43 mmol) in DMF (2 mL) was added 4-bromo-l -methylpiperidine (207 mg , 1.17 mmol). The mixture was stirred at 50°C for 16 hours. The reaction was diluted with water (50 mL), extracted with ethyl acetate (3 x 40 mL), dried over Na2SC>4, filtered and concentrated. The residue was purified by prep-TLC (dichloromethane/MeOH, 15:1] to give the crude product which was purified by prep-HPLC to afford the title compound as a white solid. MS calcd. for C29H33CIF4N6O2: 608.2; Found: 609.0 [M + H] +; XH NMR (400 MHz, MeOH- 4): 57.77 (dd, J = 6.8, 2.8 Hz, 1H), 7.63 (s, 1H), 7.55 (s, 1H), 7.44-7.38 (m, 1H), 7.14 (t, J = 8.8 Hz, 1H), 4.15-4.05 (m, 1H), 3.66 (s, 3H), 3.19-3.14 (m, 1H), 2.97-2.88 (m, 2H), 2.53-2.42 (m, 2H), 2.27 (s, 3H), 2.25-2.10 (m, 6H), 2.12-1.94 (m, 4H), 1.86-1.74 (m, 4H) ppm.
Intermediate 26

4-Bromo-l-(pyrrolidin-l-ylmethyl)-3-(trifluoromethyl)-lH-pyrazole (R-l):
To a solution of 4-bromo-3-(trifluoromethyl)-lH-pyrazole (65.0 g, 303.7 mmol) in EtOH (300 mL) was added pyrrolidine (21.6 g, 303.7 mmol) and HCHO (44.0 g, 542.7 mmol). The
reaction was stirred overnight at room temperature. The solution was concentrated in vacuo to afford the title compound as a yellow solid. MS calcd. for C9H11 B1F3N3: 297; Found: 298 [M + 1]+.
Example 154

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3-(trifluoromethyl)-lH-pyrazol-4- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. To a solution of 4- bromo-l-(pyrrolidin-l-ylmethyl)-3-(trifluoromethyl)-lH-pyrazole (6.0 g, 20.2 mmol) in dry ether (45 mL) was added t-BuLi (1.3 M, 15.5 mL, 20.2 mmol) dropwise. The reaction was stirred for 8 mins at -78 °C under an Ar atmosphere. A solution of N-(3-chloro-4- fhiorophenyl)-l-methyl-4-(5-oxo- 1,3a, 4,5,6, 6a-hexahydropentalen-2-yl)-lH-imidazole-5- carboxamide (600 mg, 1.60 mmol) in dry THF (5 mL) was dropwise added at -78 °C. The reaction was stirred for 2 h at -78 °C under an Ar atmosphere. The reaction was quenched with NH4CI solution (40 mL) and extracted with EtOAc (20 mLx3). The combined organic layer was dried over anhydrous Na2SC>4, filtered, and concentrated in vacuo. The residue was purified by prep-TLC (DCM/MeOH, 15:1) and reverse phase column chromatography to afford the title compound as a white solid. MS calcd. for C23H22CIF4N5O2: 511.1; Found: 511.9 [M+H]+. !H NMR (400 MHz, cfc-DMSO): 5 13.25 (s, 1H), 10.23 (s, 1H), 7.96 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.78 (s, 1H), 7.65 (s, 1H), 7.60-7.54 (m, 1H), 7.41 (t, 7= 9.0 Hz, 1H), 4.83 (s, 1H), 3.67 (s, 3H), 3.28-3.16 (m, 1H), 2.48-2.41 (m, 2H), 2.15-2.03 (m, 4H), 1.95-1.79 (m, 4H) ppm.
Intermediate 27

4-Hydroxycyclohexyl 4-methylbenzenesulfonate. A solution of cyclohexane- 1,4- diol (2.0 g, 1.0 eq, 17.24 mmol) in dry dichloromethane (80 mL) was added triethylamine (8.7 g, 5.0 eq, 86.21 mmol), TsCl (6.6 g, 2.0 eq, 34.48 mmol) and DMAP (42 mg, 0.02 eq, 0.34 mmol). The reaction mixture was stirred for 2 d at room temperature. The mixture was then washed with NaHCCL solution (30 mLx2) and brine (30 mL). The organic layer was dried with anhydrous Na2SC>4 and concentrated in vacuo. The residue was purified by reverse-phase column chromatography to afford 4-hydroxycyclohexyl 4- methylbenzenesulfonate as a light yellow solid.
Example 155

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-((ls,4s)-4-hydroxycyclohexyl)-3- (trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3- (trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide (160 mg, 1.0 eq, 0.31 mmol) in DMF (7 mL) was added 4-hydroxycyclohexyl 4-methylbenzenesulfonate (423 mg, 5.0 eq, 1.57 mmol) and CS2CO3 (112 mg, 1.1 eq, 0.34 mmol). The reaction mixture was stirred overnight at 100 °C in a sealed tube. Ethyl acetate (10 mL) and the organic solution washed with LiCl solution (5 mL x 2) and brine (5 mL). The organic layer was dried with anhydrous Na2SC>4 and concentrated in vacuo. The residue was purified by prep-TLC (dichloromethane/MeOH, 15:1 and ethyl acetate/MeOH, 10:1) to
afford the title compound as a white solid. MS calcd. for C29H32CIF4N5O3: 609.2; Found: 592.0 [M-18+l]+;
 NMR (400 MHz, DMSO-d6): 5 10.21 (s, 1H), 7.93 (dd, J = 6.8, 2.4 Hz, 1H), 7.77 (s, 1H), 7.62 (s, 1H), 7.57-7.51 (m, 1H), 7.38 (t, J = 9.0 Hz, 1H), 4.83 (s, 1H), 4.46 (d, J = 2.8 Hz, 1H), 4.17-4.06 (m, 1H), 3.79 (d, J = 2.0 Hz, 1H), 3.64 (s, 3H), 3.24-3.15 (m, 1H), 2.45-2.39 (m, 2H), 2.13-2.00 (m, 6H), 1.90-1.75 (m, 4H), 1.72-1.62 (m, 4H), 1.56-1.45 (m, 2H) ppm.
NMR (400 MHz, DMSO-d6): 5 10.21 (s, 1H), 7.93 (dd, J = 6.8, 2.4 Hz, 1H), 7.77 (s, 1H), 7.62 (s, 1H), 7.57-7.51 (m, 1H), 7.38 (t, J = 9.0 Hz, 1H), 4.83 (s, 1H), 4.46 (d, J = 2.8 Hz, 1H), 4.17-4.06 (m, 1H), 3.79 (d, J = 2.0 Hz, 1H), 3.64 (s, 3H), 3.24-3.15 (m, 1H), 2.45-2.39 (m, 2H), 2.13-2.00 (m, 6H), 1.90-1.75 (m, 4H), 1.72-1.62 (m, 4H), 1.56-1.45 (m, 2H) ppm.
Intermediate 28

(ls,4s)-4-Hydroxycyclohexyl 4-methylbenzenesulfonate. To a solution of (Is, 4s)- cyclohexane-l,4-diol (2.0 g, 1.0 eq, 17.24 mmol) in dry dichloromethane (80 mL) was added triethylamine (8.7 g, 5.0 eq, 86.21 mmol), TsCl (4.9 g, 1.5 eq, 25.86 mmol) and DMAP (42 mg, 0.02 eq, 0.34 mmol). The reaction mixture was stirred for 2 d at room temperature. The mixture was then washed with NaHCOa solution (30 mL x 2) and brine (30 mL). The organic layer was dried with anhydrous Na2SC>4 and concentrated in vacuo. The residue was used in the next step without further purification.
Intermediate 29

(ls,4s)-4-(Tosyloxy)cyclohexyl benzoate. To a solution of ((ls,4s)-4- hydroxycyclohexyl 4-methylbenzenesulfonate) (3.0 g, 1.0 eq, 11.11 mmol) in dry dichloromethane (50 mL) was added triethylamine (5.6 g, 5.0 eq, 55.56 mmol) and benzoyl chloride (3.1 g, 2.0 eq, 22.22 mmol). The reaction mixture was stirred overnight at room temperature. The mixture was then washed with NaHCOa solution (30 mL x 2) and brine (30 mL). The organic layer was dried with anhydrous NaaSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 10/l-to-5/l) to afford the title compound as a white solid.

(lr,4r)-4-(4-(5-(5-((3-Chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol- 4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)cyclohexyl benzoate. To a solution N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3-(trifluoromethyl)- lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- (800 mg, 1.0 eq, 1.57 mmol) in DMF (30 mL) was added (ls,4s)-4-(tosyloxy)cyclohexyl benzoate (1.2 g, 2.0 eq, 3.13 mmol) and CS2CO3 (1.0 g, 2.0 eq, 3.13 mmol). The reaction mixture was stirred overnight at 100 °C in a sealed tube. To the mixture was added ethyl acetate (30 mL) and the solution washed with LiCl solution (10 mL x 2) and brine (10 mL). The organic layer was dried with anhydrous Na2SC>4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (dichloromethane/MeOH, 30:1 - 25:1) to afford the title compound as a light yellow solid.

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-((lr,4r)-4-hydroxycyclohexyl)-3- (trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of (lr,4r)-4-(4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l- methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH- pyrazol-l-yl)cyclohexyl benzoate (620 mg, 1.0 eq, 0.87 mmol) in MeOH (30 mL) was added LiOH-fLO (365 mg, 10.0 eq, 8.70 mmol). The reaction mixture was stirred overnight at room temperature. The mixture was adjusted to pH = 7 with 6N HC1 solution and extracted with ethyl acetate (30 mL x 2). The combined organic layer was washed with NaHCOa solution (10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SC>4 and concentrated in vacuo. The residue was purified by prep-TLC (dichloromethane/MeOH = 15/1 and ethyl acetate/MeOH = 10/1) to afford the title compound as a white solid. MS calcd. for C29H32CIF4N5O3: 609.2; Found: 609.9 [M+H]+.
 NMR (400 MHz, <76-DMSO): 5 10.22 (s, 1H), 7.93 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.79 (s, 1H), 7.62 (s, 1H), 7.57-7.51 (m, 1H), 7.38 (t, 7 = 9.0 Hz, 1H), 4.84 (s, 1H), 4.65 (d, 7 = 4.0 Hz, 1H), 4.16-4.05 (m, 1H), 3.64 (s, 3H), 3.50- 3.39 (m, 1H), 3.25-3.13 (m, 1H), 2.45-2.38 (m, 2H), 2.11-1.99 (m, 4H), 1.95-1.67 (m, 10H), 1.34-1.19 (m, 2H) ppm.
NMR (400 MHz, <76-DMSO): 5 10.22 (s, 1H), 7.93 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.79 (s, 1H), 7.62 (s, 1H), 7.57-7.51 (m, 1H), 7.38 (t, 7 = 9.0 Hz, 1H), 4.84 (s, 1H), 4.65 (d, 7 = 4.0 Hz, 1H), 4.16-4.05 (m, 1H), 3.64 (s, 3H), 3.50- 3.39 (m, 1H), 3.25-3.13 (m, 1H), 2.45-2.38 (m, 2H), 2.11-1.99 (m, 4H), 1.95-1.67 (m, 10H), 1.34-1.19 (m, 2H) ppm.
Intermediate 31
 l-(2-(Tetrahydro-2H-pyran-2-yloxy)ethyl)-lH-pyrazole-3-carbaldehyde. To a solution of lH-pyrazole-5-carbaldehyde (4500.0 mg, 46.88 mmol) in DMF (25 ml) was added 2-(2-bromoethoxy)tetrahydro-2H-pyran (9750.0 mg, 46.88 mmol) and NaH (1238.0 mg, 51.57 mmol). The reaction was stirred for 16 hours at room temperature then quenched with H2O (75 ml) and extracted with EtOAc (50 ml x 3). The combined organic layers were washed with brine (50 mL), dried over sodium sulfate, and concentrated to dryness. The residue was purified by column chromatography using 0-7% methanol in dichloromethane to afford the title compound as a yellow oil. TLC: 7% MeOH/dichloromethane (/?/: 0.4), MS calcd. for C11H16N2O3: 224.3; Found: 225.0 [M+ 1] +.
l-(2-(Tetrahydro-2H-pyran-2-yloxy)ethyl)-lH-pyrazole-3-carbaldehyde. To a solution of lH-pyrazole-5-carbaldehyde (4500.0 mg, 46.88 mmol) in DMF (25 ml) was added 2-(2-bromoethoxy)tetrahydro-2H-pyran (9750.0 mg, 46.88 mmol) and NaH (1238.0 mg, 51.57 mmol). The reaction was stirred for 16 hours at room temperature then quenched with H2O (75 ml) and extracted with EtOAc (50 ml x 3). The combined organic layers were washed with brine (50 mL), dried over sodium sulfate, and concentrated to dryness. The residue was purified by column chromatography using 0-7% methanol in dichloromethane to afford the title compound as a yellow oil. TLC: 7% MeOH/dichloromethane (/?/: 0.4), MS calcd. for C11H16N2O3: 224.3; Found: 225.0 [M+ 1] +.
Intermediate 32

3-(Difluoromethyl)-l-(2-(tetrahydro-2H-pyran-2-yloxy)ethyl)-lH-pyrazole. To a solution of l-(2-(tetrahydro-2H-pyran-2-yloxy)ethyl)-lH-pyrazole-3-carbaldehyde (8000.0 mg, 35.71 mmol) in dichloromethane(50 ml) was added DAST (11500.0 mg, 71.43 mmol). The reaction stirred for 16 hours in room temperature then quenched with H2O (10 ml) in 0°C.The solvent was removed to give the crude product, which was purified by column chromatography using 0-30% ethyl acetate in petroleum ether to afford the title compound as a yellow oil. TLC: 30% ethyl acetate/petroleum ether (/?/: 0.4). MS calcd. for C11H16F2N2O2: 246.3; Found: 247.0 [M+ 1]+.

2-(3-(Difluoromethyl)-lH-pyrazol-l-yl)ethanol. To a solution of 3- (difhioromethyl)-l-(2-(tetrahydro-2H-pyran-2-yloxy)ethyl)-lH-pyrazole (4000.0 mg, 16.26 mmol) in THF/H2O (30 ml,v/v=2/l) was added HCl(aq.) until pH 3. The reaction stirred for 16 hours at room temperature. H2O (50 mL) was added and the mixture extracted with ethyl acetate (30 ml x 3). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate, and concentrated to dryness. The residue was purified by column chromatography using 0-20% methanol in dichloromethane to afford the title compound as a white solid. TLC: 20% MeOH/dichloromethane (Rf. 0.5), MS calcd. for C6H8F2N2O: 162.1; Found: 163.0 [M+ 1] +.

N-(3-Chloro-4-fluorophenyl)-4-(5-(3-(difluoromethyl)-l-(2-hydroxyethyl)-lH- pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of 2-(3-(difluoromethyl)-lH-pyrazol-l-yl)ethanol (1300.0 mg, 8.0 mmol) in THF (50 mL) was added s-BuLi (12 mL) in -78°C and the mixture stirred for 1 hour . To the reaction mixture was added N-(3-chloro-4-fluorophenyl)-l-methyl-4-(5- oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (300 mg, 0.8 mmol) and stirring continued for 1 hour. The reaction was quenched with NH4CI (aq.) and extracted with EtOAc
(30 ml x 3). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate, and concentrated to dryness, The residue was purified by column chromatography using 0-10% methanol in dichloromethane and prep-HPLC to afford the title compound as a yellow solid. TLC: 10% CHsOH/dichloromelhane (/?/: 0.5), MS calcd. for C25H27CIF3N5O3: 537.96; Found: 538.0 [M + 1] +.
 NMR (400 MHz, DMSO-tfo): 5 10.22 (s, 1H), 7.97 (dd, J = 6.8, 2.4 Hz, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.42 (t, J = 8.8 Hz, 1H), 7.02 (t, J = 54.8 Hz, 1H), 6.33 (s, 1H), 5.42 (s, 1H), 4.95 (t, J = 5.6 Hz, 1H), 4.37 (t, J = 6.4 Hz, 2H), 3.80-3.75 (m, 2H), 3.67 (s, 3H), 3.26-3.23 (m, 1H), 2.50-2.49 (m, 2H), 2.28-2.22 (m, 2H), 2.13-2.07 (m, 2H), 1.92-1.87 (m, 4H) ppm.
NMR (400 MHz, DMSO-tfo): 5 10.22 (s, 1H), 7.97 (dd, J = 6.8, 2.4 Hz, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.42 (t, J = 8.8 Hz, 1H), 7.02 (t, J = 54.8 Hz, 1H), 6.33 (s, 1H), 5.42 (s, 1H), 4.95 (t, J = 5.6 Hz, 1H), 4.37 (t, J = 6.4 Hz, 2H), 3.80-3.75 (m, 2H), 3.67 (s, 3H), 3.26-3.23 (m, 1H), 2.50-2.49 (m, 2H), 2.28-2.22 (m, 2H), 2.13-2.07 (m, 2H), 1.92-1.87 (m, 4H) ppm.
Intermediate 34
 tert-Butyl 3-((4-bromo-3-(trifluoromethyl)-lH-pyrazol-l-yl)methyl)azetidine-l- carboxylate. To a solution of 4-bromo-3-(trifluoromethyl)-lH-pyrazole (2.24 g, 10.5 mmol), CS2CO3 (2.24 g, 21.0 mmol) in DMF (15 mL) was added tert-butyl 3-(iodomethyl)azetidine- 1-carboxylate (3.74 g , 12.6 mmol). The mixture was stirred at room temperature for 16 hours. The reaction was then diluted with water (50 mL), extracted with ethyl acetate (3 x 40 mL), dried over Na2SC>4, filtered and concentrated to give the crude product, which was purified by column chromatography (0-30% ethyl acetate in petroleum ether) to afford the title compound as a white solid. TLC: 10% ethyl acetate/petroleum ether (Rr. 0.3) MS calcd. for Ci3Hi7BrF3N3O2: 383.0; Found: 328.0 [M - 56 + H] +-
tert-Butyl 3-((4-bromo-3-(trifluoromethyl)-lH-pyrazol-l-yl)methyl)azetidine-l- carboxylate. To a solution of 4-bromo-3-(trifluoromethyl)-lH-pyrazole (2.24 g, 10.5 mmol), CS2CO3 (2.24 g, 21.0 mmol) in DMF (15 mL) was added tert-butyl 3-(iodomethyl)azetidine- 1-carboxylate (3.74 g , 12.6 mmol). The mixture was stirred at room temperature for 16 hours. The reaction was then diluted with water (50 mL), extracted with ethyl acetate (3 x 40 mL), dried over Na2SC>4, filtered and concentrated to give the crude product, which was purified by column chromatography (0-30% ethyl acetate in petroleum ether) to afford the title compound as a white solid. TLC: 10% ethyl acetate/petroleum ether (Rr. 0.3) MS calcd. for Ci3Hi7BrF3N3O2: 383.0; Found: 328.0 [M - 56 + H] +-
Intermediate 35
 l-(Azetidin-3-ylmethyl)-4-bromo-3-(trifluoromethyl)-lH-pyrazole. To a mixture of tert-butyl 3-((4-bromo-3-(trifluoromethyl)-lH-pyrazol-l-yl)methyl)azetidine-l- carboxylate (3.0 g, 9.09 mmol) in dichloromethane (10 mL) was added TFA (2 mL), and the reaction stirred at room temperature for 16 hours. The reaction mixture was adjusted to pH 8 using NaHCO3 solution, then extracted with ethyl acetate (40 mL x 3), dried over Na3SO4, filtered, and concentrated to give the crude product, which was purified by reversed column chromatography to give the title compound as a white solid. MS calcd. for CsH9BrF3N3: 283.0; Found: 284.1 [M + 1]+.
l-(Azetidin-3-ylmethyl)-4-bromo-3-(trifluoromethyl)-lH-pyrazole. To a mixture of tert-butyl 3-((4-bromo-3-(trifluoromethyl)-lH-pyrazol-l-yl)methyl)azetidine-l- carboxylate (3.0 g, 9.09 mmol) in dichloromethane (10 mL) was added TFA (2 mL), and the reaction stirred at room temperature for 16 hours. The reaction mixture was adjusted to pH 8 using NaHCO3 solution, then extracted with ethyl acetate (40 mL x 3), dried over Na3SO4, filtered, and concentrated to give the crude product, which was purified by reversed column chromatography to give the title compound as a white solid. MS calcd. for CsH9BrF3N3: 283.0; Found: 284.1 [M + 1]+.
Intermediate 36

4-Bromo-l-((l-methylazetidin-3-yl)methyl)-3-(trifluoromethyl)-lH-pyrazole. A mixture of l-(azetidin-3-ylmethyl)-4-bromo-3-(trifluoromethyl)-lH-pyrazole (1.4 g, 4.9 mmol) and HCHO (37 % in water, 3.97 g, 49.0 mmol) in MeOH (5 mL) was stirred at room temperature for 8 hours. NaBH(OAc)3 (2.10 g, 9.9 mmol) was then added dropwise over 5 min and the reaction mixture stirred at room temperature for 2 hours. The reaction mixture was quenched with NaHCO3 solution (30 mL) and extracted with ethyl acetate (3 x 20 mL), dried over anhydrous Na3SO4, filtered and concentrated to give the crude product, which was purified by reverse phase column chromatography to afford the title compound as a yellow oil. MS calcd. for C9HiiBrF3N3: 297.0; Found: 298.0 [M + 1]+.
Example 158

N -(3-Chlor o-4-fluorophenyl)-4-(5 -hydroxy-5 -(1 -((1 -methylazetidin-3-yl)methyl)- 3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of 4-bromo-l-((l-methylazetidin-3-yl)methyl)-3- (trifluoromethyl)-lH-pyrazole (505 mg, 1.7 mmol) in diethyl ether (2 mL) was added t- butyllithium (2.0 mL, 2.6 mmol, 1.3 M) dropwise at -78°C, the mixture was stirred for 2 min at -78 °C in an Ar atmosphere. A solution of N-(3-chloro-4-fluorophenyl)-l-methyl-4-(5- oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (65 mg, 0.17 mmol) in anhydrous tetrahydrofuran (2 mL) was added dropwise at -78 °C. The reaction mixture was stirred for 1 h at -78 °C under an Ar atmosphere. The reaction was quenched with saturated aqueous ammonium chloride (30 mL) and extracted with ethyl acetate (3 x 20 mL). The combined organic layer was dried with anhydrous Na2SC>4 and concentrated in vacuo. The residue was purified by column chromatography (dichloromethane/MeOH=15/l) and reverse phase column chromatography to afford the title compound as a white solid. MS calcd. for C28H31CIF4N6O2: 594.2; Found: 595.0 [M+H] +.
 NMR (400 MHz, MeOH- 4): 57.86 (dd, J = 6.4, 2.4 Hz, 1H), 7.70 (brs, 1H),7.64 (s, 1H), 7.53-7.47 (m, 1H), 7.23 (t, J = 8.8 Hz, 1H), 4.27 (d, J = 7.2 Hz, 2H), 3.75 (s, 3H), 3.44-3.36 (m, 2H), 3.35-3.31 (m, 1H), 3.11-3.02 (m, 2H), 2.96-2.85 (m, 1H), 2.62-2.48 (m, 2H), 2.38-2.28 (m, 5H), 2.27-2.18 (m, 2H), 1.96-1.80 (m, 4H) ppm.
Intermediate 37
NMR (400 MHz, MeOH- 4): 57.86 (dd, J = 6.4, 2.4 Hz, 1H), 7.70 (brs, 1H),7.64 (s, 1H), 7.53-7.47 (m, 1H), 7.23 (t, J = 8.8 Hz, 1H), 4.27 (d, J = 7.2 Hz, 2H), 3.75 (s, 3H), 3.44-3.36 (m, 2H), 3.35-3.31 (m, 1H), 3.11-3.02 (m, 2H), 2.96-2.85 (m, 1H), 2.62-2.48 (m, 2H), 2.38-2.28 (m, 5H), 2.27-2.18 (m, 2H), 1.96-1.80 (m, 4H) ppm.
Intermediate 37

Allyl 3-methylenepyrrolidine-l-carboxylate. A solution of tert-butyl 3- methylenepyrrolidine-1 -carboxylate (1.83 g, 10.0 mmol) in dichloromethane (50 mL) was treated with TFA (10 mL). The reaction mixture was stirred at room temperature overnight and concentrated in vacuo to provide 3-methylenepyrrolidine. To a solution of crude 3- methylenepyrrolidine in dichloromethane (50 mL) was added allyl carbonochloridate (1.32 g, 11.0 mmol) and triethylamine (3.03 g, 30.0 mmol). The mixture was stirred at room temperature overnight then concentrated in vacuo. The residue was purified by chromatography on silica gel (eluting with petroleum ether/ethyl acetate = 20/1 (v/v)) to provide the title compound as a clear oil.
Intermediate 38 -Alloc

Allyl l-oxa-5-azaspiro[2.4]heptane-5-carboxylate. To a solution of allyl 3- methylenepyrrolidine-1 -carboxylate (1.67g, 10.0 mmol) in dichloromethane (150 mL) was added m-CPBA (1.72 g, 10.0 mmol). The mixture was stirred at room temperature overnight. To the reaction was added 100 mL saturated NaHCO and the mixture stirred for 10 min. The dichloromethane phase was separated and dried over MgSC for 10 min. The dichloromethane phase was filtered through Celite to remove MgSCU. The filtrate was concentrated in vacuo to provide crude product as a residue which was used for the next step directly.
Intermediate 39 -Alloc

Allyl 3-((4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4- yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)methyl)-3- hydroxypyrrolidine-l-carboxylate. To a solution of crude allyl l-oxa-5- azaspiro[2.4]heptane-5-carboxylate in acetonitrile was added compound N-(3-chloro-4- fluorophenyl)-4-(5-hydroxy-5-(3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)- l-methyl-lH-imidazole-5-carboxamide (1.1 g, 0.22 mmol) and potassium carbonate powder (1.36 g, 10 mmol). The mixture was heated to 75°C and stirred overnight. After cooling, the mixture was filtered through Celite to remove potassium carbonate. The filtrate was concentrated in vacuo and the residue purified by pre-HPLC to provide the title compound as white solid. MS calcd. for C32H35CIF4N6O5: 678.2; Found: 679.3 [M+l]+.

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-((3-hydroxypyrrolidin-3- yl)methyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH- imidazole-5 -carboxamide. To a solution of allyl 3-((4-(5-(5-((3-chloro-4- fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)methyl)-3-hydroxypyrrolidine-l-carboxylate ( 200 mg, 0.3 mmol) in THF (20 mL) was added Pd(PPh3)4 (20 mg ) and NaBFU powder (50 mg). The mixture was stirred at room temperature overnight. The reaction mixture was acidified using IN HC1 to pH 1.0 and the crude product used for the next step as a mixture of diastereomers. MS calcd. for C28H31CIF4N6O3: 610.2; Found: 611.3 [M+l] +.

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-((3-hydroxy-l-methylpyrrolidin-3- yl)methyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-
imidazole-5 -carboxamide Isomer I. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5- hydroxy-5-(l-((3-hydroxypyrrolidin-3-yl)methyl)-3-(trifluoromethyl)-lH-pyrazol-4- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide was treated with HCHO (30% H2O, 2 mL) and stirred at room temperature overnight. To this solution was added NaBtU powder (100 mg) and the reaction stirred for 20 min. The mixture was purified by prep-HPLC and chiral HPLC to provide N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-((3- hydroxy- 1 -methylpyrrolidin-3 -yl)methyl)-3 -(trifluoromethyl)- 1 H-pyrazol-4- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide Isomer I and Isomer IL Isomer I; MS calcd. for C29H33CIF4N6O3: 624.22; Found: 607.0 [M+-18+l]+.
 NMR (400 MHz, CD3OD): 57.86 (dd, 7 = 6.8, 2.8 Hz, 1H), 7.69 (s, 1H), 7.65 (s, 1H), 7.51-7.50 (m, 1H), 7.23 (t, 7 = 9.2 Hz, 1H), 4.21 (s, 2H), 3.76 (s, 3H), 3.33-3.31 (m, 1H), 2.82-2.73 (m, 2H), 2.59-2.57 (m, 3H), 2.44 (d, 7 = 10.8, 1H), 2.46-2.43 (m, 5H), 2.36-2.21 (m, 2H), 2.09- 2.02 (m, 1H), 1.95-1.88 (m, 4H), 1.77-1.70 (m, 1H) ppm.
NMR (400 MHz, CD3OD): 57.86 (dd, 7 = 6.8, 2.8 Hz, 1H), 7.69 (s, 1H), 7.65 (s, 1H), 7.51-7.50 (m, 1H), 7.23 (t, 7 = 9.2 Hz, 1H), 4.21 (s, 2H), 3.76 (s, 3H), 3.33-3.31 (m, 1H), 2.82-2.73 (m, 2H), 2.59-2.57 (m, 3H), 2.44 (d, 7 = 10.8, 1H), 2.46-2.43 (m, 5H), 2.36-2.21 (m, 2H), 2.09- 2.02 (m, 1H), 1.95-1.88 (m, 4H), 1.77-1.70 (m, 1H) ppm.

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-((3-hydroxy-l-methylpyrrolidin- 3-yl)methyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH- imidazole-5 -carboxamide Isomer II. MS calcd. for C29H33CIF4N6O3: 624.22; Found: 607.0 [M-18+l]+. NMR (400 MHz, CD3OD): 57.86 (dd, 7 = 6.8, 2.8 Hz, 1H), 7.69 (s, 1H), 7.65 (s, 1H), 7.50-7.49 (m, 1H), 7.23 (t, 7 = 9.2 Hz, 1H), 4.21 (s, 2H), 3.76 (s, 3H), 3.33-3.30 (m, 1H), 2.84-2.78 (m, 2H), 2.64-2.57 (m, 3H), 2.47 (d, 7 = 10.8 Hz, 1H), 2.37-2.31 (m, 5H), 2.25-2.22 (m, 2H), 2.10-2.03 (m, 1H), 1.94-1.85 (m, 4H), 1.79-1.72 (m, 1H) ppm.

Ethyl 2-(4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4- yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)-2- methylpropanoate. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3- (trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide (80.0 mg, 0.15 mmol) in DMF (3 mL) was added ethyl 2-bromo-2- methylpropanoate (91.0 mg, 0.47 mmol), CS2CO3 (250.0 mg, 0.78 mmol) and Nal (45.0 mg, 0.3 mmol). The reaction was stirred at 50°C overnight. Water was added and the solution extracted with ethyl acetate, dried over Na2SC>4 then concentrated to give the crude product, which was purified by column chromatography to afford the title compound as a brown solid. TLC: 7% MeOH/dichloromethane (/?/: 0.6), MS calcd. for C29H32CIF4N5O4: 625.2; Found: 625.9 [M + 1]+.

4-(5-(l-(l-Amino-2-methyl-l-oxopropan-2-yl)-3-(trifluoromethyl)-lH-pyrazol-4- yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-
imidazole-5 -carboxamide. A solution of ethyl 2-(4-(5-(5-((3-chloro-4- fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3- (trifhioromethyl)-lH-pyrazol-l-yl)-2-methylpropanoate (60.0 mg, 0.1 mmol) and NH3/CH3OH (3 mF) was stirred at 50°C overnight. The solvent was removed under reduced pressure to give the crude product, which was purified by column chromatography and prep- HPLC to the title compound as a white solid. TLC: 9% MeOH/dichloromethane (Rf. 0.2), MS calcd. for C27H29CIF4N6O3: 597.0; Found: 579.0 [M -18 + 1]+.
 NMR (400 MHz, DMSO- d6): 5 10.25 (s, 1H), 7.96 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.79 (s, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, 7 = 9.2 Hz, 1H), 7.27 (s, 1H), 7.05 (s, 1H), 4.86 (s, 1H), 3.67 (s, 3H), 3.50-3.43 (m, 2H), 3.25-3.19 (m, 1H), 2.16-2.06 (m, 4H), 1.93-1.82 (m, 4H), 1.70 (s, 6H) ppm.
NMR (400 MHz, DMSO- d6): 5 10.25 (s, 1H), 7.96 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.79 (s, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, 7 = 9.2 Hz, 1H), 7.27 (s, 1H), 7.05 (s, 1H), 4.86 (s, 1H), 3.67 (s, 3H), 3.50-3.43 (m, 2H), 3.25-3.19 (m, 1H), 2.16-2.06 (m, 4H), 1.93-1.82 (m, 4H), 1.70 (s, 6H) ppm.
Example 163

N-(3-Chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-(4-oxocyclohexyl)-3- (trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide. (CP-AIA-1422-6): To a solution of N-(3-chloro-4-fluorophenyl)-4-(5- hydroxy-5-(l-(4-hydroxycyclohexyl)-3-(trifluoromethyl)-lH-pyrazol-4- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (560 mg, 0.92 mmol) in acetone (15 mL) was added Dess-Martin Periodinane (1.17 g, 2.76 mmol) and Na2CO , and the mixture stirred at 65 °C for 30 min. The reaction was quenched with Na2SO (aq). The suspension was extracted with ethyl acetate. The combined organic layers were dried and concentrated in vacuo and purified by column chromatography on silica gel with 9-18 % of MeOH in dichloromethane to afford the title compound as a white solid (230 mg, 41.0 %) TLC: 10% MeOH/dichloromethane (Rf: 0.4); MS calcd. for C29H30CIF4N5O3: 607.2; Found: 590.0 [M - 18 + l]+.

N -(3-Chlor o-4-fluorophenyl)-4-(5 -hydroxy-5 -(1 -(4-hydroxy-4-methylcyclohexyl)- 3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide Isomer I. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(l-(4- oxocyclohexyl)-3-(trifluoromethyl)- lH-pyrazol-4-yl)octahydropentalen-2-yl)- 1-methyl- 1H- imidazole-5-carboxamide (230 mg, 0.38 mmol) in dry THF (10 mL) at 65 °C was added 3.0 M (hexane) CH^Mgl (2.5 mL, 7.6 mmol) slowly and the reaction stirred for 2 hours. The reaction was quenched with H2O and the suspension extracted with ethyl acetate (3x25 mL). The combined organic layers were dried and concentrated in vacuo to give the crude product which was purified by reversed phase prep-HPLC and chiral-SFC to afford N-(3-Chloro-4- fhiorophenyl)-4-(5-hydroxy-5-(l-(4-hydroxy-4-methylcyclohexyl)-3-(trifluoromethyl)-lH- pyrazol-4-yl)octahydropentalen-2-yl)-l -methyl- lH-imidazole-5-carboxamide Isomer I and Isomer II. Isomer I; TLC: 10% MeOH/dichloromethane (Rf. 0.5); MS calcd. for C30H34CIF4N5O3: 624.1; Found: 606
 NMR (400 MHz, DMSO-cfc): 5 10.23 (s, 1H), 7.96 (dd, J = 6.8, 2.4 Hz, 1H), 7.79 (s, 1H), 7.65 (s, 1H), 7.59-7.53 (m, 1H), 7.41 (t, J = 9.2 Hz, 1H), 4.84 (s, 1H), 4.21 (s, 1H),4.O9 (brs, 1H), 3.67 (s, 3H), 3.26-3.20 (m, 1H), 2.41- 2.33 (m, 2H), 2.13-2.02 (m, 6H), 1.92-1.61 (m, 10H), 1.13 (s, 3H) ppm.
NMR (400 MHz, DMSO-cfc): 5 10.23 (s, 1H), 7.96 (dd, J = 6.8, 2.4 Hz, 1H), 7.79 (s, 1H), 7.65 (s, 1H), 7.59-7.53 (m, 1H), 7.41 (t, J = 9.2 Hz, 1H), 4.84 (s, 1H), 4.21 (s, 1H),4.O9 (brs, 1H), 3.67 (s, 3H), 3.26-3.20 (m, 1H), 2.41- 2.33 (m, 2H), 2.13-2.02 (m, 6H), 1.92-1.61 (m, 10H), 1.13 (s, 3H) ppm.

N -(3-Chlor o-4-fluorophenyl)-4-(5 -hydroxy-5 -(1 -(4-hydroxy-4-methylcyclohexyl)- 3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide Isomer II. TLC: 10% MeOH/dichloromethane (/?/: 0.5); MS calcd. for C30H34CIF4N5O3: 623.2; Found: 606 [M- 18 +
 NMR (400 MHz, DMSO-tfo): 5 10.22 (s, 1H), 7.96 (dd, J = 6.8, 2.4 Hz, 1H), 7.87 (s, 1H), 7.65 (s, 1H), 7.59-7.56 (m, 1H), 7.41 (t, J = 8.8 Hz, 1H), 4.84 (s, 1H), 4.42 (s, 1H), 3.33 (brs, 1H), 3.67 (s, 3H), 3.27-3.22 (m, 2H), 2.51-2.49 (m, 2H), 2.13-2.06 (m, 4H), 1.91-1.80 (m, 8H), 1.61-1.51 (m, 4H), 1.19 (s, 3H) ppm.
NMR (400 MHz, DMSO-tfo): 5 10.22 (s, 1H), 7.96 (dd, J = 6.8, 2.4 Hz, 1H), 7.87 (s, 1H), 7.65 (s, 1H), 7.59-7.56 (m, 1H), 7.41 (t, J = 8.8 Hz, 1H), 4.84 (s, 1H), 4.42 (s, 1H), 3.33 (brs, 1H), 3.67 (s, 3H), 3.27-3.22 (m, 2H), 2.51-2.49 (m, 2H), 2.13-2.06 (m, 4H), 1.91-1.80 (m, 8H), 1.61-1.51 (m, 4H), 1.19 (s, 3H) ppm.
Intermediate 41

(lr,4r)-Methyl 4-(tosyloxy)cyclohexanecarboxylate. To a solution of (lr,4r)-methyl 4-hydroxycyclohexanecarboxylate (1.0 g, 6.3 mmol) in dichloromethane (15 mL) was added TsCl (3.0 g, 15.8 mmol), NEt3 (1.9 g, 18.9 mmol) and DMAP (154 mg, 1.3 mmol). The mixture was stirred at 25 °C overnight. The solvent was removed under reduce pressure to give the crude product, which was purified by silica gel column chromatography to afford the title compound as a white solid. TLC: 11% ethyl acetate/petroleum ether (Rf: 0.4); MS calcd. for C15H20O5S: 312.1; Found: 313.3 [M + 1]+.

Methyl (ls,4s)-4-(4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH- imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l- yl)cyclohexane-l-carboxylate. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy- 5-(3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide ( 200 mg, 0.4 mmol) in DMF (5 ml) was added (lr,4r)-methyl 4- (tosyloxy)cyclohexanecarboxylate (370 mg, 1.2 mmol) and CS2CO3 (140 mg, 0.4 mmol). The mixture was stirred at 50°C overnight. Water (50 ml) was added, and the mixture extracted with ethyl acetate. The organic layer was dried and concentrated. The crude product was purified by prep-HPLC to afford the title compound as a white solid. TLC: 9% MeOH/dichloromethane (/?/: 0.5); MS calcd. for C31H34CIF4N5O4: 651.2; Found: 652.3 [M + 1]+. !H NMR (400 MHz, MeOH-d4): 7.88-7.86 (dd, J = 6.8, 2.4 Hz, 1H), 7.68 (d, J = 0.8 Hz, 1H), 7.65 (s, 1H), 7.52-7.48 (m, 1H), 7.26-7.21 (t, J = 9.2 Hz, 1H), 4.20-4.18 (m, 1H), 3.76 (s, 3H), 3.70 (s, 3H), 3.34-3.28 (m, 1H), 2.70-2.68 (m, 1H), 2.57-2.54 (m, 2H), 2.35-2.30 (m, 2H), 2.27-2.17 (m, 4H), 2.00-1.86 (m, 8H), 1.77-1.73 (m, 2H) ppm.

(ls,4s)-4-(4-(5-(5-((3-Chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-
4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l-
yl)cyclohexane-l-carboxylic acid. To a solution of methyl (ls,4s)-4-(4-(5-(5-((3-chloro-4- fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)cyclohexane-l-carboxylate ( 30 mg, 0.05 mmol) in THF (1 mL) and H2O (0.5 mL) was added NaOH aq. (3 M, 1.5 mL). The mixture was stirred at room temperature overnight. HC1 aq. (IM) was added dropwise to reach pH < 4. Water (20 mL) was added, and the solution extracted with ethyl acetate. The organic layer was dried and concentrated under reduced pressure to give the title compound as colorless oil. TLC: 12% MeOH/dichloromethane (Rf. 0.4). MS calcd. for C30H32CIF4N5O4: 637.2; Found: 638.3 [M + 1] +-

4-(5-(l-((ls,4s)-4-Carbamoylcyclohexyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of (ls,4s)-4-(4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l- methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH- pyrazol-l-yl)cyclohexane-l-carboxylic acid ( 25 mg, 0.04 mmol) in THF(3 mL) was added NH4CI (10 mg, 0.2 mmol) and diisopropyl-ethylamine (40 mg, 0.31 mmol). The mixture was stirred at room temperature for 0.5 h then HATU (44 mg, 0.12 mmol) added and stirring continued overnight. Water (20 ml) was added, and the mixture extracted with ethyl acetate. The organic layer was dried and concentrated under reduced pressure. The crude product was purified by prep-HPLC to afford the title compound as a white solid. TLC: 5% MeOH/dichloromethane (Rf. 0.3). MS calcd. for C30H33CIF4N6O3: 636.2; Found: 619.0 [M - 18 + 1]+. ' H-NMR (400 MHz, MeOD): 7.89-7.86 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.70 (d, 7 = 0.4 Hz, 1H), 7.65 (s, 1H), 7.52-7.48 (m, 1H), 7.26-7.21 (m, 1H), 4.26-4.21 (m, 1H), 3.76 (s, 3H), 3.35-3.30 (m, 1H), 2.58-2.51 (m, 3H), 2.37-2.31 (m, 2H), 2.27-2.19 (m, 4H), 2.03-1.86 (m, 8H), 1.76-1.71 (m, 2H) ppm.
Intermediate 43

(ls,4s)-Methyl 4-(tosyloxy)cyclohexanecarboxylate. To a solution of methyl (ls,4s)-4-hydroxycyclohexane-l-carboxylate (2.0 g, 12.66 mmol) in dichloromethane (100 mL) was added DMAP (1.5g, 12.66 mmol), EI3N (2.8 g,31.65 mmol) and TsCl (3.6 g, 18.99 mmol) and the reaction stirred for 16 hours at room temperature. The solvent was removed under reduced pressure and the crude product was purified by column chromatography using 0-20% EtOAc in petroleum ether to afford the title compound as a white solid. TLC: 25% ethyl acetate/petroleum ether (Rf. 0.4), MS calcd. for C15H20O5S: 312.1; Found: 330.1 [M + 18]+.

Methyl (lr,4r)-4-(4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH- imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l- yl)cyclohexane-l-carboxylate. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy- 5-(3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide (250.0 mg, 0.49 mmol) in DMF (15 ml) was added (ls,4s)-methyl 4- (tosyloxy)cyclohexanecarboxylate (610.0 mg, 1.96 mmol) and K2CO3 (74.0 mg,0.54mmol) and the reaction heated to 100°C and stirred for 16 hours .The reaction mixture was cooled to room temperature and quenched with H2O (50 ml) and, extracted with EtOAc (30 ml x 3). The combined organic layers were washed with brine (30mL), dried over sodium sulfate, and
concentrated to dryness. The resulting residue was purified by column chromatography using 0-10% methanol in dichloromethane to afford the title compound as a white solid. TLC: 7% MeOH/dichloromethane (Rf. 0.4). MS calcd. for C31H34CIF4N5O4: 651.2; Found: 634.0 [M - 18 + 1]+. NMR (400 MHz, MeOH-d4): 57.88 (dd, J = 6.8, 2.4 Hz, 1H), 7.70 (s, 1H), 7.64 (s, 1H), 7.51-7.48 (m, 1H), 7.25 (t, J = 8.8 Hz, 1H), 4.17-4.13 (m, 1H), 3.75 (s, 3H), 3.67 (s, 3H), 3.34-3.31 (m, 1H), 3.30-3.29 (m, 2H), 2.59-2.56 (m, 2H), 2.44-2.40 (m, 1H), 2.35-2.30 (m, 2H), 2.24-2.22 (m, 2H), 2.13-2.10 (m, 4H), 1.93-1.83 (m, 6H), 1.62-1.58 (m, 2H) ppm.

(lr,4r)-4-(4-(5-(5-((3-Chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol- 4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l- yl)cyclohexane-l-carboxylic acid. To a solution of methyl (lr,4r)-4-(4-(5-(5-((3-chloro-4- fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)cyclohexane-l-carboxylate (30.0 mg, 0.045 mmol) in THF/H2O = 2/1 (v/v) (15 ml) was added NaOH (30 mg, 0.75 mmol). The reaction stirred for 16 hours in room temperature then acidified with HC1 (aq.) to pH 4. The solution was extracted with EtOAc (30 ml x 3). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate, and concentrated to dryness. The resulting residue was purified by column chromatography using 0-20% methanol in dichloromethane to afford the title compound as a white solid. TLC: 20% MeOH/dichloromethane (/?/: 0.4). MS calcd. for C30H32CIF4N5O4: 637.2; Found: 619.8 [M - 18 + 1]+ .

4-(5-(l-((lr,4r)-4-Carbamoylcyclohexyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide To a solution of (lr,4r)-4-(4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l- methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH- pyrazol-l-yl)cyclohexane-l-carboxylic acid (25.0 mg, 0.039mmol) in DMF (5 mL) was added HATU (44.46 mg, 0.117 mmol), NH4CI (3.2 mg, 0.0585 mmol) and diisopropylethylamine (15.09mg, 0.117 mmol). The mixture was stirred for 4 h at room temperature then quenched with H2O (20 ml) and extracted with EtOAc (10 ml x 3). The combined organic layers were washed with brine (10 mL), dried over sodium sulfate, and concentrated to dryness, The residue was purified by column chromatography using 0-20% methanol in dichloromethane and prep-HPLC to afford the title compound as a white solid. TLC: 20% MeOH/dichloromethane (/?/: 0.4). MS calcd. for C30H33CIF4N6O3: 636.2; Found: 619.0 [M - 18 + 1]+ ; !H NMR (400 MHz, DMSO-tfo): 5 10.23 (s, 1H), 7.97 (dd, J = 6.8, 2.8 Hz, 1H), 7.83 (s, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.43 (t, J = 9.2 Hz, 1H), 7.27 (s, 1H), 6.75 (s,lH), 4.86 (s, 1H), 4.16-4.12 (m, 1H), 3.67 (s, 3H), 3.22-3.16 (m, 1H), 2.49-2.46 (m, 2H), 2.13-1.99 (m, 7H), 1.91-1.79 (m, 6H), 1.73-1.69 (m, 2H), 1.50-1.46 (m, 2H) ppm.
Intermediate 45

( Ls,3s)-Methyl 3-(tosyloxy)cyclobutanecarboxylate To a solution of (Is, 3s) -methyl 3 -hydroxycyclobutanecarboxylate (1.0 g, 15.4 mmol), triethylamine (3.1 g, 30.8 mmol) and DMAP (188 mg, 1.5 mmol) in dichloromethane (30 mL) was added TosCl (2.5 g, 23.1
mmol). The mixture was stirred at room temperature for 16 h. The reaction was treated with NaHCCL solution (50 mL) and extracted with dichloromethane (3 x 50 mL), the combined organic layers were dried with anhydrous Na2SC>4 and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether/ethyl acetate = 3/1 (v/v)) to afford oil (3.0 g, 68.6%). MS calcd. for CI3HI6O5S: 284.1; Found: 302.2 [M+18] +.

Methyl (lr,3r)-3-(4-(5-(5-((3-chloro-4-fhiorophenyl)carbamoyl)-l-methyl-lH- imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3-(trifluoromethyl)-lH-pyrazol-l- yl)cyclobutane-l-carboxylate. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy- 5-(3-(trifluoromethyl)-lH-pyrazol-4-yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide (250 mg , 0.49 mmol) and CS2CO3 (175 mg, 0.54 mmol) in DMF (2.5 mL) was added (Is, 3s) -methyl 3-(tosyloxy)cyclobutanecarboxylate (417 mg, 1.47 mmol). The mixture was stirred for 16 hours at 50 °C. The reaction was treated with water (50 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layer was washed with brine, dried, and concentrated in vacuo. The residue was purified by column chromatography (dichloromethane/MeOH = 15/1 (v/v)) to afford the title compound as a white solid. MS calcd. for C29H30CIF4N5O4: 623.2; Found: 624.1 [M+H]+.
 NMR (400 MHz, MeOH-d4): 5 7.87 (dd, J = 6.4, 2.4 Hz, 1H), 7.72 (d, J = 1.2 Hz, 1H), 7.65 (s, 1H), 7.53-7.47 (m, 1H), 7.23 (t, J = 8.8 Hz, 1H), 5.08-4.98 (m, 1H), 3.76 (s, 3H), 3.73 (s, 3H), 3.37-3.32 (m, 1H), 3.25- 3.16 (m, 1H), 2.90-2.78 (m, 2H), 2.75-2.65 (m, 2H), 2.62-2.50 (m, 2H), 2.38-2.18 (m, 4H), 1.96-1.82 (m, 4 H) ppm.
Example 171
NMR (400 MHz, MeOH-d4): 5 7.87 (dd, J = 6.4, 2.4 Hz, 1H), 7.72 (d, J = 1.2 Hz, 1H), 7.65 (s, 1H), 7.53-7.47 (m, 1H), 7.23 (t, J = 8.8 Hz, 1H), 5.08-4.98 (m, 1H), 3.76 (s, 3H), 3.73 (s, 3H), 3.37-3.32 (m, 1H), 3.25- 3.16 (m, 1H), 2.90-2.78 (m, 2H), 2.75-2.65 (m, 2H), 2.62-2.50 (m, 2H), 2.38-2.18 (m, 4H), 1.96-1.82 (m, 4 H) ppm.
Example 171

4-(5-(l-((lr,3r)-3-Carbamoylcyclobutyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)-5- hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH-imidazole-5- carboxamide. To a solution of methyl (lr,3r)-3-(4-(5-(5-((3-chloro-4- fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3- (trifluoromethyl)- lH-pyrazol-l-yl)cyclobutane-l-carboxylate (50 mg , 0.08 mmol) was added NH3 in MeOH (2 mL).The mixture was stirred at 50 °C for 24 hours. The reaction mixture was concentrated in vacuo. The residue was purified by reversed phase column chromatography afford the title compound. MS calcd. for C28H29CIF4N6O3: 608.2; Found: 609.2 [M+H]+. !H NMR (400 MHz, MeOH-d4): 57.87 (dd, J = 6.8, 2.8 Hz, 1H), 7.73 (s 1H), 7.65 (s, 1 H), 7.53-7.46 (m, 1H), 7.23 (t, J = 8.8 Hz, 1H), 5.07-4.98 (m, 1H), 3.76 (s, 3H), 3.37-3.32 (m, 1 H), 3.22-3.12 (m, 1H), 2.84-2.73 (m, 2H), 2.71-2.62 (m, 2H), 2.60-2.50 (m, 2H), 2.37-2.18 (m, 4H), 1.95-1.80 (m, 4 H) ppm.
Intermediate 46

4-Bromo-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole-3-carbaldehyde A 100 mL flask was charged with 4-bromo-lH-pyrazole-3-carbaldehyde (3 g, 17 mmol) and THF (40 mL), added NaH (1 g, 26 mmol) slowly at 0 °C, the reaction mixture was stirred at 0 °C for 0.5 h. Then added 2-(trimethylsilyl)ethoxymethyl chloride (3.6 mL, 20 mmol), stirred at
room temperature overnight. The mixture was quenched with H2O (20 mL) and extracted with ethyl acetate (30 ml x 3), combined organic layer was dried over Na2SC>4, filtered, concentrated, and purified by silica gel column chromatography (petroleum ether: ethyl acetate = 60 : 1, then petroleum ether: ethyl acetate = 30: 1) to afford the title compound as a colorless oil. TLC: petroleum ether/ethyl acetate = 10/1 (v/v), Rf = 0.4.
Intermediate 47

4-Bromo-3-(difluoromethyl)-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole. A 100 mL flask was charged with 4-bromo-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole- 3-carbaldehyde (3.07 g, 10 mmol) and dichloromethane (30 mL). DAST (5.5 mL, 40 mmol) was added slowly at 0 °C and the reaction warmed to room temperature slowly and stirred for 6 h. The mixture was extracted with ethyl acetate (30 ml x 3). The combined organic layer was dried over Na2SC>4, filtered, concentrated, and purified by silica gel column chromatography to afford the title compound as a colorless oil. TLC: dichloromethane, Rf = 0.9.
Intermediate 48

4-Bromo-3-(difluoromethyl)-lH-pyrazole A 100 mL flask was charged with 4- bromo-3-(difhioromethyl)-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole (2.7 g, 8.3 mmol) and CFaCOOH/dichloromethane (10 mL/20 mL). The mixture was stirred at room temperature for 4 h. The pH was adjusted to 8.0 with NaHCO solution. The mixture was extracted with ethyl acetate (30 ml x 3). The combined organic layer was dried over Na2SC>4, filtered, and concentrated to afford the title compound as a white solid.
Intermediate 49

4-Bromo-3-(difluoromethyl)-l-(pyrrolidin-l-ylmethyl)-lH-pyrazole. A 100 mL flask was charged with 4-bromo-3-(difluoromethyl)-lH-pyrazole (1.6 g, 8.2 mmol), HCHO (674 mg, 9 mmol), pyrrolidine (640 mg, 9 mmol) and EtOH (25 mL). The mixture was stirred overnight at room temperature. The mixture was extracted with ethyl acetate (30 ml x 3) and the combined organic layer dried over Na2SC>4, filtered, and concentrated to afford the title compound as a colorless oil.
Example 172

N-(3-Chloro-4-fluorophenyl)-4-(5-(3-(difluoromethyl)-lH-pyrazol-4-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. A 100 mL flask was charged with 4-bromo-3-(difluoromethyl)-l-(pyrrolidin-l-ylmethyl)-lH-pyrazole (1.76 g, 6.3 mmol) and THF (40 mL). n-BuLi (2.5 mL, 6.3 mmol) slowly added at - 78 °C under an Argon atmosphere and the mixture stirred at - 78 °C for 2 min. N-(3-Chloro-4-fluorophenyl)- l-methyl-4-(5-oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (300 mg, 0.8 mmol) was then added at - 78 °C. The mixture was stirred at - 78 °C for 3 h under Argon. The mixture was quenched with H2O (20 mL) and extracted with ethyl acetate (30 ml x 3). The combined organic layer was dried over Na2SC>4, filtered, concentrated, and purified by prep- HPLC to afford the title compound as a white solid. TLC: dichloromethane/MeOH = 10/1, Rf = 0.5. MS calcd. for C23H23CIF3N5O2: 493.2; Found: 494 [M + 1]+.
Example 173

Ethyl 2-(4-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4- yl)-2-hydroxyoctahydropentalen-2-yl)-3-(difluoromethyl)-lH-pyrazol-l-yl)-2- methylpropanoate. A 100 mL flask was charged with N-(3-chloro-4-fluorophenyl)-4-(5-(3- (difluoromethyl)- 1 H-pyrazol-4-yl)-5 -hydroxy octahydropentalen-2-yl)- 1 -methyl- 1 H- imidazole-5-carboxamide (150 mg, 0.3 mmol), ethyl 2-bromo-2-methylpropanoate (60 mg, 1.8 mmol), CS2CO3 (90 mg, 0.3 mmol) and DMF (20 mL). The mixture was warmed up to 50 °C and stirred overnight. The reaction mixture was extracted with ethyl acetate (30 ml x 3). The combined organic layer was washed with H2O three times. The organic layer was dried over Na2SC>4, filtered, concentrated and the crude product purified by prep-HPLC to afford the title compound as a white solid. TLC: dichloromethane/MeOH = 10/1 (v/v), Rf = 0.6. MS calcd. for C29H33CIF3N5O4: 607.22; Found: 590 [M - 18+l]+.
 NMR (400 MHz, DMSO- d6): 5 10.24 (s, 1H), 7.96 (dd, J = 6.8 Hz, 2.8 Hz, 1H), 7.79 (s, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, J = 9.2 Hz, 1H), 7.14 (t, J = 54.4 Hz, 1H), 5.06 (s, 1H), 4.09 (q, J = 6.8 Hz, 2H), 3.67 (s, 3H), 3.27-3.20 (m, 1H), 2.50-2.49 (m, 2H), 2.12-2.07 (m, 4H), 1.92-1.79 (m, 4H), 1.74 (s, 6H), 1.11 (t, J = 7.2 Hz, 3H) ppm.
Example 174
NMR (400 MHz, DMSO- d6): 5 10.24 (s, 1H), 7.96 (dd, J = 6.8 Hz, 2.8 Hz, 1H), 7.79 (s, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, J = 9.2 Hz, 1H), 7.14 (t, J = 54.4 Hz, 1H), 5.06 (s, 1H), 4.09 (q, J = 6.8 Hz, 2H), 3.67 (s, 3H), 3.27-3.20 (m, 1H), 2.50-2.49 (m, 2H), 2.12-2.07 (m, 4H), 1.92-1.79 (m, 4H), 1.74 (s, 6H), 1.11 (t, J = 7.2 Hz, 3H) ppm.
Example 174

2-(4-(5-(5-((3-Chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2- hydroxyoctahydropentalen-2-yl)-3-(difluoromethyl)-lH-pyrazol-l-yl)-2- methylpropanoic acid. A 50 mL flask was charged with ethyl 2-(4-(5-(5-((3-chloro-4- fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3- (difluoromethyl)-lH-pyrazol-l-yl)-2-methylpropanoate (30 mg, 0.05 mmol), NaOH (8 mg, 0.20 mmol) and MeOH (10 mL), The mixture was stirred at room temperature overnight. The pH value was adjusted to 8.0 with HC1 (IM). The mixture was extracted with ethyl acetate (30 ml x 3). The combined organic layer was dried over Na2SC>4, filtered, and concentrated. The residue was purified by prep-HPLC to afford the title compound as a white solid. MS calcd. for C27H29CIF3N5O4: 579.19; Found: 580 [M + 1]+.
 NMR (400 MHz, DMSO-d6): 5 10.24 (s, 1H), 7.96 (dd, J = 6.8 Hz, 2.4 Hz, 1H), 7.71 (s, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, J = 8.8 Hz, 1H), 7.13 (t, J = 54.4 Hz, 1H), 5.01 (s, 1H), 3.67 (s, 3H), 3.28-3.21 (m,
NMR (400 MHz, DMSO-d6): 5 10.24 (s, 1H), 7.96 (dd, J = 6.8 Hz, 2.4 Hz, 1H), 7.71 (s, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, J = 8.8 Hz, 1H), 7.13 (t, J = 54.4 Hz, 1H), 5.01 (s, 1H), 3.67 (s, 3H), 3.28-3.21 (m,
1H), 2.51-2.50 (m, 2H), 2.10-2.06 (m, 4H), 1.94-1.80 (m, 4H), 1.67 (s, 6H) ppm.
Example 175

4-(5-(l-(l-Amino-2-methyl-l-oxopropan-2-yl)-3-(difluoromethyl)-lH-pyrazol-4- yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3-chloro-4-fluorophenyl)-l-methyl-lH- imidazole-5 -carboxamide. To a mixture of 2-(4-(5-(5-((3-chloro-4- fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-3- (difluoromethyl)-lH-pyrazol-l-yl)-2-methylpropanoic acid (15 mg, 0.03 mmol), NH4CI (7 mg, 0.15 mmol), diisopropyl ethylamine (15 mg, 0.12 mmol) and DMF (4 mL) was added HATU (49 mg, 0.13 mmol) the reaction stirred at room temperature for 5 h. The mixture was extracted with ethyl acetate (30 ml x 3). The combined organic layer was dried over Na2SC>4, filtered, concentrated then purified by prep-HPLC to afford the title compound as a white solid. MS calcd. for C27H30CIF3N6O3: 578.20; Found: 561 [M - 18 + 1]+.
 NMR (400 MHz, DMSO-d6): 5 10.24 (s, 1H), 7.96 (dd, J = 6.4 Hz, 2.4 Hz, 1H), 7.69 (s, 1H), 7.65(s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, J = 9.2 Hz, 1H), 7.23 (s 1H), 7.14 (t, J = 54.4 Hz, 1H), 6.89 (s, 1H), 5.01 (s, 1H), 3.67 (s, 3H), 3.26-3.20 (m, 1H), 2.50-2.43 (m, 2H), 2.12-2.07 (m, 4H), 1.93-1.79 (m, 4H), 1.68 (s, 6H) ppm.
NMR (400 MHz, DMSO-d6): 5 10.24 (s, 1H), 7.96 (dd, J = 6.4 Hz, 2.4 Hz, 1H), 7.69 (s, 1H), 7.65(s, 1H), 7.59-7.55 (m, 1H), 7.41 (t, J = 9.2 Hz, 1H), 7.23 (s 1H), 7.14 (t, J = 54.4 Hz, 1H), 6.89 (s, 1H), 5.01 (s, 1H), 3.67 (s, 3H), 3.26-3.20 (m, 1H), 2.50-2.43 (m, 2H), 2.12-2.07 (m, 4H), 1.93-1.79 (m, 4H), 1.68 (s, 6H) ppm.

Ethyl 2-((5-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l-methyl-lH-imidazol-4- yl)-2-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-pyrazol-3-yl)oxy)-2-fluoroacetate.
To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3-hydroxy-l-methyl-lH- pyrazol-5-yl)octahydropentalen-2-yl)-l -methyl- lH-imidazole-5-carboxamide (450.2 mg , 0.95 mmol) in dry DMF (10 mL) was added cesium carbonate (619.1 mg, 1.9 mmol). Ethyl 2-bromo-2-fluoroacetate (192.4 mg, 1.04 mmol) in DMF (5 mL) was slowly added at 0°C and the reaction mixture stirred at 0°C overnight under a N2-atmosphere. After completion, the reaction mixture was quenched with ice cold water (100 mL) and extracted with EtOAc (3x50 mL). The combined organic layer was dried over anhydrous Na2SC>4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (eluting with CH3OH/DCM, 1:13 (v/v)) to provide the title compound as a yellow solid. TLC: 5% MeOH/DCM (R 0.5). MS calcd. for C27H30CIF2N5O5: 577.2; Found: 559.9 [M- 18+1]+
Example 176

N-(3-Chloro-4-fluorophenyl)-4-(5-(3-(l-fluoro-2-hydroxy-2-methylpropoxy)-l- methyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide Isomer I. Methyl magnesium bromide (2.5M in THF, 0.6 mL, 1.8 mmol) was slowly added to a solution of ethyl 2-((5-(5-(5-((3-chloro-4-fluorophenyl)carbamoyl)-l- methyl-lH-imidazol-4-yl)-2-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-pyrazol-3- yl)oxy)-2-fluoroacetate (202.3 mg, 0.35 mmol) in dry THF (10 mF) at 0 °C under a N2- atmosphere. The reaction mixture was stirred at room temperature for 10 mins. After completion, the reaction mixture was quenched with saturated NH4CI (50 mL) and extracted with EtOAc (3x20mL). The combined organic layer was dried over anhydrous Na2SC>4, filtered and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (eluting with CH3OH/DCM, 1:13 (v/v)) followed by chromatographic chiral separation to provide N-(3-chloro-4-fluorophenyl)-4-(5-(3-(l-fluoro-2-hydroxy-2- methylpropoxy)- 1-methyl- lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)- 1-methyl- 1H- imidazole-5-carboxamide Isomer I and Isomer II. Isomer I: TLC: 5% MeOH/DCM (RJ: 0.4). MS calcd. for C27H32CIF2N5O4: 563.21; Found: 564.0 [M+l]+, 546.0 [M-18+l]+. ' H-NMR (400 MHz, DMSO-<76): 5 10.13 (s, 1H), 7.96 (dd, 7 = 6.8, 2.8 Hz, 1H), 7.65 (s, 1H), 7.58- 7.56 (m, 1H), 7.40 (t, 7 = 8.8 Hz, 1H), 5.73 (s, 1H), 5.59 (d, 7 = 61.2 Hz, 1H), 5.29 (s, 1H), 4.94 (s, 1H), 3.77 (s, 3H), 3.72 (s, 3H), 3.28-3.22 (m, 1H), 2.50-2.24 (m, 2H, merged), 2.20- 2.07 (m, 4H), 1.86 (m, 4H), 1.14 (s , 6H) ppm.
Example 177

N-(3-Chloro-4-fluorophenyl)-4-(5-(3-(l-fluoro-2-hydroxy-2-methylpropoxy)-l- methyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5- carboxamide Isomer II. TLC: 5% MeOH/DCM (Rfi 0.4). MS calcd. for C27H32CIF2N5O4: 563.2; Found: 564.0 [M+l]+, 546.0 [M-18+l]+. ^-NMR (400 MHz, DMSO-tfc): 5 10.17 (s, 1H), 7.96 (dd, 7 = 6.8, 2.8 Hz, 1H), 7.65 (s, 1H), 7.59-7.55 (m, 1H), 7.40 (t, 7 = 8.8 Hz, 1H), 5.73 (s, 1H), 5.70 (d, 7 = 61.2 Hz, 1H), 5.26 (s, 1H), 4.92 (s, 1H), 3.77 (s, 3H), 3.68 (s, 3H), 3.28-3.22 (m, 1H), 2.50-2.24 (m, 2H, merged), 2.21-2.08 (m, 4H), 1.89-1.84 (m, 4H), 1.14 (s , 6H) ppm.

N-(3-Chloro-4-fluorophenyl)-4-(5-(3-(difluoromethoxy)-l-methyl-lH-pyrazol-5- yl)-5-hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. A solution of N-(3-chloro-4-fluorophenyl)-4-(5-hydroxy-5-(3-hydroxy-l-methyl-lH-pyrazol-5- yl)octahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (199.1 mg , 0.42 mmol) in dry DMF (3 mF) was added DBU (127.9 mg, 0.84 mmol) and ethyl 2-bromo-2,2-
difluoroacetate (127.9 mg, 0.63 mmol) at 0°C. The reaction mixture was stirred at 70°C overnight under a N2-atmosphere. The progress of reaction was monitored by LCMS. After completion, the reaction mixture was quenched with ice cold water (30 mL) and extracted with EtOAc (3x10 mL). The combined organic layer was dried over anhydrous Na2SC>4, filtered and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (eluting with CH3OH/DCM, 1:15 v/v ) followed by prep-HPLC to provide the title compound as a white solid. TLC: 5% MeOH/DCM (RJ: 0.6). MS calcd. for C24H25CIF3N5O3: 523.16; Found: 524.2 [M+l]+, 506.2 [M-18+l]+.
 NMR (400 MHz, DMSO-<76): 5 7.88 (dd, J = 6.8, 2.8 Hz, 1H), 7.65 (s, 1H), 7.51-7.48 (m, 1H), 7.23 (t, J =9.2 Hz, 1H), 6.85 (t, J =74 Hz, 1H), 5.84 (s, 1H), 3.87 (s, 3H), 3.76 (s, 3H), 3.33-3.31 (m, 1H), 2.59-2.57 (m, 2H), 2.39-2.34 (m, 2H), 2.27-2.24 (m, 2H), 1.98-1.87 (m, 4H) ppm.
NMR (400 MHz, DMSO-<76): 5 7.88 (dd, J = 6.8, 2.8 Hz, 1H), 7.65 (s, 1H), 7.51-7.48 (m, 1H), 7.23 (t, J =9.2 Hz, 1H), 6.85 (t, J =74 Hz, 1H), 5.84 (s, 1H), 3.87 (s, 3H), 3.76 (s, 3H), 3.33-3.31 (m, 1H), 2.59-2.57 (m, 2H), 2.39-2.34 (m, 2H), 2.27-2.24 (m, 2H), 1.98-1.87 (m, 4H) ppm.

N-(3-Chloro-4-fluorophenyl)-4-(5-(l-ethyl-3-nitro-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. To a solution of l-ethyl-3-nitro-lH-pyrazole (1.0 mg, 7.09 mmol) in 100 mL of THF was added LDA(7 mL, 11.5 mmol). After addition, the reaction was stirred at -78 °C for 0.5 h. N-(3-Chloro-4- fluorophenyl)-l-methyl-4-(5-oxooctahydropentalen-2-yl)-lH-imidazole-5-carboxamide (324.4 mg, 0.88 mmol) was added and the reaction mixture warmed to room temperature and stirred for another 2 h. The reaction was quenched with water and extracted with ethyl acetate (100 mL x 3). The combined organic layer was dried over anhydrous Na2SC>4, concentrated, and purified by prep-HPLC to afford the title compound as a yellow solid.

N-(3-Cyano-4-fluorophenyl)-4-(5-(l-ethyl-3-nitro-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide. To a solution of N-(3-chloro-4-fluorophenyl)-4-(5-(l-ethyl-3-nitro-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (100.0 mg, 0.194 mmol ) in dioxane(10 ml) and H2O(5 ml) was added Zn(CN)2 (114 mg, 0.968 mmol), t- BuXPhos (42 mg, 0.097 mmol ), and 3rd generation t-BuXPhos-pre-catalyst (78 mg, 0.097 mmol). The reaction was stirred at 60 °C overnight. The reaction was quenched with water and extracted with ethyl acetate (10 mL x 3). The combined organic layer was dried over anhydrous Na2SC>4, concentrated, and purified by CC to afford the title compound as a white solid.

4-(5-(3-Amino-l-ethyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3- cyano-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide. To a solution of N-(3- cyano-4-fluorophenyl)-4-(5-(l-ethyl-3-nitro-lH-pyrazol-5-yl)-5- hydroxyoctahydropentalen-2-yl)-l-methyl-lH-imidazole-5-carboxamide (50.0 mg, 0.1 mmol ) in EtOH(l ml) was added SnCh (60 mg, 0.27 mmol) and the solution stirred at 85
°C for 1 h. The reaction was quenched with saturated NaOH (aq), concentrated, and purified by column chromatography (DCM/MeOH, 30:1 (v/v)) to afford the title compound as a white solid.

4-(5-(3-Amino-4-chloro-l-ethyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2- yl)-N-(3-cyano-4-fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide. To a solution of 4-(5-(3-amino-l-ethyl-lH-pyrazol-5-yl)-5-hydroxyoctahydropentalen-2-yl)-N-(3-cyano-4- fluorophenyl)-l-methyl-lH-imidazole-5-carboxamide (30.0 mg, 0.06 mmol) in dichloromethane (2 ml), was added N-chlorosuccinimide (10 mg, 0.072 mmol) and the reaction stirred at room temperature for 2 h. The reaction was quenched with water and extracted with CH2CI2 (10 mL x 3). The combined organic layer was concentrated and purified by prep-HPLC to afford the title compound as a white solid. MS calcd. for C25H27CIFN7O2: 511; Found: 512 [
 10.36 (s, 1H), 8.17 (dd, J = 5.6 Hz, 2.8 Hz, 1H), 7.95-7.91 (m, 1H), 7.66 (s, 1H), 7.53 (t, J = 9.2 Hz, 1H), 5.31 (s, 1H), 4.59 (s, 2H), 4.11 (dd,J=7.2,6.8 Hz, 2H), 3.68 (s, 3H), 3.27-3.22 (m, 1H), 2.39- 2.34 (m, 2H), 2.10-1.92 (m, 8H)), 1.25 (t, J=6.85 Hz, 3H) ppm.
10.36 (s, 1H), 8.17 (dd, J = 5.6 Hz, 2.8 Hz, 1H), 7.95-7.91 (m, 1H), 7.66 (s, 1H), 7.53 (t, J = 9.2 Hz, 1H), 5.31 (s, 1H), 4.59 (s, 2H), 4.11 (dd,J=7.2,6.8 Hz, 2H), 3.68 (s, 3H), 3.27-3.22 (m, 1H), 2.39- 2.34 (m, 2H), 2.10-1.92 (m, 8H)), 1.25 (t, J=6.85 Hz, 3H) ppm.
Table 5. Examples 183-302 were synthesized according to the methods described elsewhere in this case




































































Biological Data
Assay Measuring Activity of Test Compounds on Viral Production from HepAD38 Cells
HepAD38 cells grown in a T-150 flask (Corning, cat#: 430825) with Growth Medium (DMEM/F12 (1:1) (Hyclone, cat#: SH30023.02), IX Pen/Strep (Invitrogen, cat#: 15140- 122), 10% FBS (Tissue Culture Biologies, cat#: 101), 250 pg/mL G418 (Alfa Aesar, cat#: J62671), Ipg/mL Tetracycline (Teknova, cat#: T3320)) were detached with 0.25% trypsin- EDTA (Invitrogen, cat#: 25200-056). Tetracycline-free treatment medium (15 mL DMEM/F12 (1:1)- lx Pen/step, with 2% FBS, Tet-system approved (Clontech, cat#: 631106) were then added to mix, transferred into a 50 ml conical tube (Falcon, cat#: 21008-918,) and spun at 1300 rpm for 5 min. Pelleted cells were then re-suspended/washed with 50 mL of IX DPBS (Invitrogen, cat#: 14190-136) 2 times and 50 mL treatment medium twice. HepAD38 cells were then re-suspended with 10 mL of treatment medium, syringed and counted. Wells of 96-well clear bottom TC plate (Coming, cat#: 3904,) were seeded at 50,000 cells/well in 180 pL of treatment medium, and 20 pL of either 10% DMSO (Sigma, cat#: D4540) as controls or a 10X solution of test compounds in 10% DMSO in treatment media was added for a final compound concentration starting at 10 pM, and plates were incubated in 5% CO2 incubator at 37°C for 5 days.
Subsequently viral load production was assayed by quantitative PCR (qPCR) of the HBV core sequence. PCR reaction mixture containing forward primers HBV-f 5'- CTGTGCCTTGGGTGGCTTT-3’ (IDT DNA), Reverse primers HBV-r 5'- AAGGAAAGAAGTCAGAAGGCAAAA-3' (IDT DNA), Fluorescent TaqMantm Probes HB V-probe 5 '-FAM/AGCTCC AAA/ZEN/TTCTTTATAAGGGTCGATGTC/3IABkFQ -3 ' (IDT DNA), 10 pL/well of PerfeCTa® qPCR ToughMix® (Quanta Biosciences, Cat#: 95114- 05K), and 6 pL/well of DEPC water (Alfa Aesar, cat#: J62087) was prepared. Four pL of supernatant was added to 16 pL of the reaction mixture in a qPCR plate (Applied Biosytems, Cat#: 4309849), sealed with a film (Applied Biosystems, Cat#: 4311971), centrifuged for a few seconds, and subsequently run on an Applied Biosystems VIIA7. The PCR mixture was incubated at 45 °C for 5 min, then 95 °C for 10 min, followed by 40 cycles of 10 seconds at 95 °C and 20 seconds at 60°C. Viral load was quantified against known HBV DNA standards by using ViiA™ 7 Software. Viral load in the supernatant from wells with treated cells were compared against viral load in supernatant from DMSO control wells (> 3 per plate). Cell viability assay was performed with CellTiter-Glo Luminescent Cell Viability Assay (Promega, cat#: G7573) with modification. Mixed appropriate amount of CellTiter-Glo (CTG) IX DPBS in a 1:1 ratio, added 100 uL of the mixture to each well followed completely removal of all supernatant in each well without touching cell surface. Incubated
the plate at room temperature for 10 min on an orbital shaker, and then read the plate with a plate reader (TECAN M1000 or Envision). EC50 or CC50 values were calculated through curve-fitting of the four-parameter nonlinear-logistic -regression model (GraphPad Prism or Dotmatics). CC50 values were all >10 |1M. Table 6 gives the viral load lowering EC50 values for exemplified compounds of the invention. In the table, A; EC50 < 10 nM, B; EC50 >10, < 100 nM and C; EC50 >100, < 500 nM.
Table 6.











VI. Stereochemistry of Examples AIA-225

5-Amino-N-(3-chloro-4-fluorophenyl)-3-(5-hydroxy-5- (methylthiomethyl)octahydropentalen-2-yl)-l-methyl-lH-pyrazole-4-carboxamide. To a solution of 5-amino-N-(3-chloro-4-fluorophenyl)-3-(hexahydro-TH-spiro[oxirane-2,2'- pentalene]-5'-yl)-l-methyl-lH-pyrazole-4-carboxamide (200 mg, 0.495 mmol) in THF/H2O (6 mL/2 mL) was added NaSMe (138.6 mg, 1.98 mmol). The mixture was stirred at rt overnight. The solvent was removed, and the crude product purified by silica gel column chromatography using 3:1 (v/v) petroleum ether/ethyl acetate to afford 5-amino-N-(3-chloro- 4-fluorophenyl)-3-(5-hydroxy-5-(methylthiomethyl)octahydropentalen-2-yl)-l-methyl-lH- pyrazole-4-carboxamide (100 mg, 44.7%) as a yellow solid. MS (m/z) calcd. for C21H26CIFN4O2S: 452; Found: 453 [M+l] +.
AIA-227-1, AIA-227-2

5-Amino-N-(3-chloro-4-fluorophenyl)-3-((2r,5r)-5-hydroxy-5- (methylsulfonylmethyl)octahydropentalen-2-yl)-l-methyl-lH-pyrazole-4-carboxamide (AIA-227-1) and 5-Amino-N-(3-chloro-4-fluorophenyl)-3-((2s,5s)-5-hydroxy-5- (methylsulfonylmethyl)octahydropentalen-2-yl)-l-methyl-lH-pyrazole-4-carboxamide (AIA-227-2). To a solution of 5-amino-N-(3-chloro-4-fluorophenyl)-3-(5-hydroxy-5- (methylthiomethyl)octahydropentalen-2-yl)- 1 -methyl- 1 H-pyrazole-4-carboxamide (100 mg, 0.22 mmol) in DCM (5 mL) was added m-CPBA (114.8 mg, 0.66 mmol). The mixture was stirred at rt overnight. The solvent was removed, and the crude material purified by silica gel column chromatography using 3:1 (v/v) DCM/MeOH to afford AIA-227 (40 mg, 37.3%) as a white solid. MS (m/z) calcd. for C21H26CIFN4O4S: 484, Found: 485 [M+l]+. AIA-227 was separated by SFC to give AIA-227-1 (4 mg) as a white solid and AIA-227-2 (4 mg) as a white solid. AIA-227-1:
 NMR (400 MHz, DMSO-tfo): 5 8.95 (s, 1H), 7.91 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.54 - 7.50 (m, 1H), 7.35 (t, 7 = 9.2 Hz, 1H), 5.97 (s, 2H), 4.79 (s, 1H), 3.59 - 3.53 (m, 1H), 3.49 (s, 3H), 3.35 (s, 2H), 2.97 (s, 3H), 2.67 - 2.60 (m, 2H), 2.18 - 2.12 (m, 2H), 2.07 - 2.02 (m, 2H), 1.45 - 1.36 (m, 4H) ppm. AIA-227-2:
NMR (400 MHz, DMSO-tfo): 5 8.95 (s, 1H), 7.91 (dd, 7 = 6.8, 2.4 Hz, 1H), 7.54 - 7.50 (m, 1H), 7.35 (t, 7 = 9.2 Hz, 1H), 5.97 (s, 2H), 4.79 (s, 1H), 3.59 - 3.53 (m, 1H), 3.49 (s, 3H), 3.35 (s, 2H), 2.97 (s, 3H), 2.67 - 2.60 (m, 2H), 2.18 - 2.12 (m, 2H), 2.07 - 2.02 (m, 2H), 1.45 - 1.36 (m, 4H) ppm. AIA-227-2:
 NMR (400 MHz, DMSO- d6) 5 8.94 (s, 1H), 7.91 (dd, 7 = 2.8, 2.4 Hz, 1H), 7.53 - 7.49 (m, 1H), 7.34 (t, 7 = 9.2 Hz, 1H), 5.97 (s, 2H), 4.87 (s, 1H), 3.49 (s, 3H), 3.43 - 3.35 (m, 1H), 3.25 (s, 2H), 2.97 (s, 3H), 2.49 (s, 2H), 2.15 - 2.09 (m, 2H), 2.02 - 1.97 (m, 2H), 1.73 - 1.60 (m, 4H) ppm.
NMR (400 MHz, DMSO- d6) 5 8.94 (s, 1H), 7.91 (dd, 7 = 2.8, 2.4 Hz, 1H), 7.53 - 7.49 (m, 1H), 7.34 (t, 7 = 9.2 Hz, 1H), 5.97 (s, 2H), 4.87 (s, 1H), 3.49 (s, 3H), 3.43 - 3.35 (m, 1H), 3.25 (s, 2H), 2.97 (s, 3H), 2.49 (s, 2H), 2.15 - 2.09 (m, 2H), 2.02 - 1.97 (m, 2H), 1.73 - 1.60 (m, 4H) ppm.
AIA-227-2

Alternative synthesis of 5-amino-N-(3-chloro-4-fluorophenyl)-3-((2s,5s)-5- hydroxy-5-(methylsulfonylmethyl)octahydropentalen-2-yl)-l-methyl-lH-pyrazole-4- carboxamide. To a solution of dimethylsulfone (77.0 g, 818.7 mmol) in THF (800 mL) was added n-BuLi (327.5 mL, 818.7 mmol, 2.5M) dropwise at -78 °C. The resulting solution was allowed to warm to -20 °C and stirred for 1 hr. The reaction was cooled to -78 °C, and a solution of AIA-002 (40.0 g, 102.3 mmol) in anhydrous tetrahydrofuran (1200 mL) was added over 2 hr. The mixture was warmed to RT and stirred for an additional 4 hr. The reaction mixture was quenched with saturated aqueous ammonium chloride solution (200 mL). The solvent was removed, followed by dilution with water, extraction with ethyl acetate (3 x 200 mL), drying over Na2SC>4, filtration, and concentration to give the crude product. The crude product was purified by column chromatography using 0-5% (v/v) methanol in DCM and basic prep-HPLC to afford 5-amino-N-(3-chloro-4-fluorophenyl)-3-((2s,5s)-5- hydroxy-5-(methylsulfonylmethyl)octahydropentalen-2-yl)-l-methyl-lH-pyrazole-4- carboxamide (26.0 g, 52.4%) as a white solid. MS (m/z) calcd. for C21H26CIFN4O4S: 484; Found: 485 [M+l]+.
 NMR (400 MHz, DMSO-tfo): 5 8.96 (s, 1H), 7.92 (dd, 7 = 6.8, 2.8 Hz, 1H), 7.54 - 7.50 (m, 1H), 7.35 (t, 7 = 8.8 Hz, 1H), 5.98 (s, 2H), 4.88 (s, 1H), 3.49 (s, 3H), 3.42 - 3.37 (m, 1H), 3.25 (s, 2H), 2.97 (s, 3H), 2.15 - 2.10 (m, 2H), 2.03 - 1.97 (m, 2H), 1.73 - 1.60 (m, 4H) ppm.
NMR (400 MHz, DMSO-tfo): 5 8.96 (s, 1H), 7.92 (dd, 7 = 6.8, 2.8 Hz, 1H), 7.54 - 7.50 (m, 1H), 7.35 (t, 7 = 8.8 Hz, 1H), 5.98 (s, 2H), 4.88 (s, 1H), 3.49 (s, 3H), 3.42 - 3.37 (m, 1H), 3.25 (s, 2H), 2.97 (s, 3H), 2.15 - 2.10 (m, 2H), 2.03 - 1.97 (m, 2H), 1.73 - 1.60 (m, 4H) ppm.
A crystal with size of 0.08 x 0.10 x 0.20mm of compound AIA-227-2 was obtained from EtOH after 20 days of volatilization and was used for X-ray diffraction data collection. The data were collected on a Bruker SMART CCD area-detector diffractometer at room temperature using CuKrz radiation by a>/<p scan mode. 10846 reflections were collected, of which 3754 reflections were unique (Rint = 0.0507).
The crystal belongs to monoclinic crystal system, with a space group P2i/c. The unit cell parameters were as follows: a= 6.6143(3), £>=14.0381(8), c=23.6870(14)A, t=y=90.0°, ^97.702(3) °, V= 2179.5(2)A3, Z=4.
The structure was solved by direct methods and all of the non-H atoms were refined against F by full-matrix least-squares methods using the SHELXTL program. All H atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms. Multi-scans absorption correction method was used, and the maximum and minimum transmission parameters were 0.7531 and 0.6017, respectively. The final R, wRi, GOF are 0.0457, 0.1293 and 1.024, respectively.
There is one C21H26FCIN4O4S molecule in the asymmetric unit and hydrogen bonds can be found between them, which play an important role for the stable packing of the crystal structure.
The ORTEP plot for compound AIA-227-2 is present in Fig. 1. The relative stereochemistry scheme of compound AIA-227-2 is shown in Fig. 2. The depictions of stereochemistry in the chemical structures of related examples are based on this assignment.
INCORPORATION BY REFERENCE
All publications and patents mentioned herein, including those items listed below, are hereby incorporated by reference in their entirety for all purposes as if each individual publication or patent was specifically and individually incorporated by reference. In case of conflict, the present application, including any definitions herein, will control.
EQUIVALENTS
While specific embodiments of the subject disclosure have been discussed, the above specification is illustrative and not restrictive. Many variations of the disclosure will become apparent to those skilled in the art upon review of this specification. The full scope of the disclosure should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
Unless otherwise indicated, all numbers expressing quantities of ingredients, reaction conditions, and so forth used in the specification and claims are to be understood as being modified in all instances by the term “about.” Accordingly, unless indicated to the contrary, the numerical parameters set forth in this specification and attached claims are
approximations that may vary depending upon the desired properties sought to be obtained by the present disclosure.
Claims
1. A compound of Formula I

, or a pharmaceutically acceptable salt thereof, wherein:
L1 is a bond, Ci-4alkylene, Ci-4alkenylene, Ci-4alkynylene, haloCi-4alkylene, hydroxyCi-4alkylene, NRcCi-4alkyl, OC alkyl, O, NRC, C(O), C(O)O, C(O)NRC, S(O)t, S(O)tNRc, S(O)tCi-4alkyl, and S(O)thaloCi-4alkyl;
L3 is Ci-ealkylene, C2-ealkenylene or C2-ealkynylene, wherein the Ci-ealkylene, C2- ealkenylene, C2-ealkynylene is optionally substituted with 1-10 substituents independently selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci- ealkoxy, haloCi-ealkoxy, hydroxyCi-ealkoxy, RaRbN-Ci-ealkoxy, and haloCi-ealkylNRc-;
X1 is NRxl, O or S;
X4 is O or S;
X5 is O, S or NR6a;
Ra, Rb and Rc are independently selected for each occurrence from the group consisting of hydrogen, C1-6 alkyl, and haloCi -ealkyl;
Rd is hydrogen, OH, C1-6 alkyl or C1-6 alkoxy;
Rxl is hydrogen, C1-4 alkyl, C1-4 alkenyl, C1-4 alkynyl, haloCi-4 alkyl, or C3-6 monocycloalkyl;
ROa is independently selected for each occurrence from the group consisting of hydrogen, halogen, OH, CN, NO2, RaRbN-, Ci-4alkyl and haloCi-4 alkyl;
R6a is hydrogen or C1-4 alkyl, haloCi-4alkyl or C ^cycloalkyl;
R6b is Ci-ealkyl, C2-ealkenyl or C2-ealkynyl, wherein the Ci-ealkyl, C2-ealkenyl, C2- ealkynyl is optionally substituted with 1-10 substituents independently selected from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi-ealkoxy, RaRbN-Ci-ealkoxy, and haloCi-ealkylNRc-;
R°, R4a, R6 and R11 are independently selected for each occurrence from the group consisting of hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl,
siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, R6b, R6bC(O)-, R6bC(O)O-, R6bC(O)NRc-, R6bS(O)tNRc-, R6bS(O)t-, R6bO-, R6bNRc-, R6bC(O)-L3-, and R6bC(O)O-L3-, R6bC(O)NRc-L3-, R6bS(O)tNRc-L3-, R6bS(O)q-L3-, R6bO-L3-, and R6bNRc-L3-;
R1 is a phenyl or 5-6 membered monocyclic heteroaryl, wherein the phenyl or 5-6 membered monocyclic heteroaryl is optionally substituted with one, two, or three independently selected R11 groups;
R2 and R8 are independently selected from the group consisting of hydrogen, halo, CN, OH, RaRbN, Ci-4alkyl, haloCi^alkyl, C -smonocycloalkyl, Ci-4alkoxy, and haloCi- 4alkoxy;



2.
2. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein X1 is XRxl and Rxl is hydrogen or methyl.
3. The compound of claim 2, or a pharmaceutically acceptable salt thereof, wherein Rxl is methyl.
4. The compound according to any one of claims 1-3, or a pharmaceutically acceptable salt thereof, wherein p is 0.
5. The compound according to any one of claims 1-4, or a pharmaceutically acceptable salt thereof, wherein R2 is hydrogen.
6. The compound according to any one of Claims 1-5, or a pharmaceutically acceptable salt thereof, wherein: R1 is
 ; R11 is independently selected for each occurrence from the group consisting of halogen, CN, Ci-ealkyl and haloCi-6 alkyl; and zl is 0, 1, 2 or 3.
; R11 is independently selected for each occurrence from the group consisting of halogen, CN, Ci-ealkyl and haloCi-6 alkyl; and zl is 0, 1, 2 or 3.
7. The compound of Claim 6, or a pharmaceutically acceptable salt thereof, wherein for each occurrence R11 is independently selected from the group consisting of CN, F, Cl, Br and I.
8. The compound of Claim 7, or a pharmaceutically acceptable salt thereof, wherein

9. The compound of Claim 7, or a pharmaceutically acceptable salt thereof, wherein

10. The compound according to any one of claims 1-9, or a pharmaceutically acceptable salt thereof, wherein

11. The compound according to any one of claims 1-10, or a pharmaceutically acceptable salt thereof, wherein

12. The compound of claim 11, or a pharmaceutically acceptable salt thereof, wherein:

R4b is selected for each occurrence from the group consisting of \ hydrogen, halogen, OH, CN, NO2, oxo, RdN=, hydrazino, formyl, azido, silyl, siloxy, HOC(O)-, RaRbN-, RaRbNS(O)t-, RaRbNC(O)-, Ci-ealkyl, C2-ealkenyl, C2-ealkynyl, haloCi-ealkyl, hydroxyCi- 6alkyl-, RaRbNCi-6alkyl-, HOC(O)Ci-6alkyl-, Ci-6alkylC(O)-, Ci-6alkylC(O)O-, Ci- ealkylC(O)NRc-, Ci-6alkylS(O)t-, Ci-6alkylS(O)tNRc-, Ci-ealkoxy, haloCi-ealkoxy, hydroxyCi- ealkoxy-, RaRbNCi-6alkoxy-, RaRbNCi-6alkylNRc-, Ci-6alkylNRaCi-6alkyleneNRc-, Ci- ealkoxyCi-ealkylene-, haloCi-ealkoxyCi-ealkylene-, Ci-ealkoxyC(O)-, Ci-6alkylS(O)tCi- ealkylene-, Ci-6alkylS(O)tNRaCi-6alkylene-, Ci-6alkylC(O)Ci-6alkylene-, Ci-6alkylC(O)OCi- ealkylene- and R9;
R9 is R12S(O)t-Ci-6alkylene-, R12S(O)tNH-Ci-6alkylene-, R12C(O)NH-Ci-6alkylene-, R12S(O)t-haloCi-6alkylene-, R12S(O)tNH-haloCi-6alkylene-, or R12C(O)NH-haloCi-6alkylene-; and
R12 is RaRbN-, Ci-ealkyl, Ci-ehaloalkyl, Ci-ealkoxy, or Ci-ehaloalkoxy.
13. The compound according to any one of claims 1-12, or a pharmaceutically acceptable salt thereof, wherein R5 is In certain embodiments, R5 is R5a, R5d or R6.
14. The compound according to any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein L1 is a bond.
15. The compound according to any one of claims 1-14, or a pharmaceutically acceptable salt thereof, wherein R8 is hydrogen, OH or C1-6 alkoxy.
16. The compound of claim 15, or a pharmaceutically acceptable salt thereof, wherein R8 is OH.
17. A pharmaceutical composition comprising the compound according to any one of claims 1-16, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
18. A method of treating Hepatitis B (HBV) infection in a subject in need thereof, the method comprising: administering to the subject a therapeutically effective amount of a compound according to any one of claims 1-16, or a pharmaceutically acceptable salt thereof.
19. A method of treating Hepatitis B (HBV) infection in a subject in need thereof, the method comprising: administering to the subject a therapeutically effective amount of pharmaceutical composition of claim 17.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163257776P | 2021-10-20 | 2021-10-20 | |
| US63/257,776 | 2021-10-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023069547A1 true WO2023069547A1 (en) | 2023-04-27 |
Family
ID=84359665
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2022/047169 WO2023069547A1 (en) | 2021-10-20 | 2022-10-19 | 5-membered heteroaryl carboxamide compounds for treatment of hbv |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023069547A1 (en) |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013006394A1 (en) | 2011-07-01 | 2013-01-10 | Institute For Hepatitis And Virus Research | Sulfamoylbenzamide derivatives as antiviral agents against hbv infection |
| WO2013010069A1 (en) | 2011-07-13 | 2013-01-17 | Indiana University Research And Technology Corporation | Modified viral structural protein with antiviral activity |
| WO2013102655A1 (en) | 2012-01-06 | 2013-07-11 | Janssen R&D Ireland | 4,4-disubstituted-1,4-dihydropyrimidines and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014033176A1 (en) | 2012-08-28 | 2014-03-06 | Janssen R&D Ireland | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014033167A1 (en) | 2012-08-28 | 2014-03-06 | Janssen R&D Ireland | Fused bicyclic sulfamoyl derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014037480A1 (en) | 2012-09-10 | 2014-03-13 | F. Hoffmann-La Roche Ag | 6-amino acid heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2014074906A1 (en) | 2012-11-09 | 2014-05-15 | Indiana University Research And Technology Corporation | Alternative uses for hbv assembly effectors |
| WO2014089296A2 (en) | 2012-12-06 | 2014-06-12 | Institute For Hepatitis And Virus Research | Functionalized benzamide derivatives as antiviral agents against hbv infection |
| WO2014106019A2 (en) | 2012-12-27 | 2014-07-03 | Philadelphia Health & Education Corporation, D/B/A Drexel | Novel antiviral agents against hbv infection |
| WO2014131847A1 (en) | 2013-02-28 | 2014-09-04 | Janssen R&D Ireland | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014161888A1 (en) | 2013-04-03 | 2014-10-09 | Janssen R&D Ireland | N-phenyl-carboxamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014184328A1 (en) | 2013-05-17 | 2014-11-20 | F. Hoffmann-La Roche Ag | 6-bridged heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2014184365A1 (en) | 2013-05-17 | 2014-11-20 | Janssen R&D Ireland | Sulphamoylthiophenamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014184350A1 (en) | 2013-05-17 | 2014-11-20 | Janssen R&D Ireland | Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2020086533A1 (en) * | 2018-10-22 | 2020-04-30 | Assembly Biosciences, Inc. | 5-membered heteroaryl carboxamide compounds for treatment of hbv |
| WO2021216656A1 (en) * | 2020-04-22 | 2021-10-28 | Assembly Biosciences, Inc. | 5-membered heteroaryl carboxamide compounds for treatment of hbv |
-
2022
- 2022-10-19 WO PCT/US2022/047169 patent/WO2023069547A1/en active Application Filing
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013006394A1 (en) | 2011-07-01 | 2013-01-10 | Institute For Hepatitis And Virus Research | Sulfamoylbenzamide derivatives as antiviral agents against hbv infection |
| WO2013010069A1 (en) | 2011-07-13 | 2013-01-17 | Indiana University Research And Technology Corporation | Modified viral structural protein with antiviral activity |
| WO2013102655A1 (en) | 2012-01-06 | 2013-07-11 | Janssen R&D Ireland | 4,4-disubstituted-1,4-dihydropyrimidines and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014033176A1 (en) | 2012-08-28 | 2014-03-06 | Janssen R&D Ireland | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014033167A1 (en) | 2012-08-28 | 2014-03-06 | Janssen R&D Ireland | Fused bicyclic sulfamoyl derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014033170A1 (en) | 2012-08-28 | 2014-03-06 | Janssen R&D Ireland | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014037480A1 (en) | 2012-09-10 | 2014-03-13 | F. Hoffmann-La Roche Ag | 6-amino acid heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2014074906A1 (en) | 2012-11-09 | 2014-05-15 | Indiana University Research And Technology Corporation | Alternative uses for hbv assembly effectors |
| WO2014089296A2 (en) | 2012-12-06 | 2014-06-12 | Institute For Hepatitis And Virus Research | Functionalized benzamide derivatives as antiviral agents against hbv infection |
| WO2014106019A2 (en) | 2012-12-27 | 2014-07-03 | Philadelphia Health & Education Corporation, D/B/A Drexel | Novel antiviral agents against hbv infection |
| WO2014131847A1 (en) | 2013-02-28 | 2014-09-04 | Janssen R&D Ireland | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014161888A1 (en) | 2013-04-03 | 2014-10-09 | Janssen R&D Ireland | N-phenyl-carboxamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014184328A1 (en) | 2013-05-17 | 2014-11-20 | F. Hoffmann-La Roche Ag | 6-bridged heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2014184365A1 (en) | 2013-05-17 | 2014-11-20 | Janssen R&D Ireland | Sulphamoylthiophenamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2014184350A1 (en) | 2013-05-17 | 2014-11-20 | Janssen R&D Ireland | Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| WO2020086533A1 (en) * | 2018-10-22 | 2020-04-30 | Assembly Biosciences, Inc. | 5-membered heteroaryl carboxamide compounds for treatment of hbv |
| WO2021216656A1 (en) * | 2020-04-22 | 2021-10-28 | Assembly Biosciences, Inc. | 5-membered heteroaryl carboxamide compounds for treatment of hbv |
Non-Patent Citations (3)
| Title |
|---|
| CARREIRAKVAERNO: "Classics in Stereoselective Synthesis", 2009, WILEY-VCH |
| RAUTIOKUMPULAINEN ET AL., NATURE REVIEWS DRUG DISCOVERY, vol. 7, 2008, pages 255 |
| WANG Y ET AL.: "Transbody against hepatitis B virus core protein inhibits hepatitis B virus replication in vitro", INT. IMMUNOPHARMACOL, 2014 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TW201840545A (en) | Cyclic sulfamide compounds and methods of using same | |
| US12030869B2 (en) | 5-membered heteroaryl carboxamide compounds for treatment of HBV | |
| US20230183213A1 (en) | 5-membered heteroaryl carboxamide compounds for treatment of hbv | |
| WO2020051319A1 (en) | Cyclic sulfamide compounds for treatment of hbv | |
| WO2023164186A1 (en) | Benzothia(dia)zepine compounds for treatment of hbv and hdv | |
| WO2020051320A1 (en) | Cyclic sulfamide compounds for treatment of hbv | |
| US20230295096A1 (en) | Pyrazole carboxamide compounds for treatment of hbv | |
| WO2023164183A1 (en) | Benzothia(dia)zepine compounds for treatment of hbv and hdv | |
| WO2021216660A1 (en) | 5-membered heteroaryl carboxamide compounds for treatment of hbv | |
| WO2023069547A1 (en) | 5-membered heteroaryl carboxamide compounds for treatment of hbv | |
| WO2023069545A1 (en) | 5-membered heteroaryl carboxamide compounds for treatment of hbv | |
| WO2023069544A1 (en) | 5-membered heteroaryl carboxamide compounds for treatment of hbv | |
| WO2021216661A1 (en) | Pyrazole carboxamide compounds for treatment of hbv | |
| WO2023164179A1 (en) | Benzothia(dia)zepine compounds for treatment of hbv and hdv | |
| WO2023164181A1 (en) | Benzothia(dia)zepine compounds for treatment of hbv and hdv | |
| WO2023067517A1 (en) | Novel crystalline forms | |
| JP2024541124A (en) | New crystal morphology |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22806069 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22806069 Country of ref document: EP Kind code of ref document: A1 |