WO2013030750A1 - Antiviral compounds - Google Patents
Antiviral compounds Download PDFInfo
- Publication number
- WO2013030750A1 WO2013030750A1 PCT/IB2012/054381 IB2012054381W WO2013030750A1 WO 2013030750 A1 WO2013030750 A1 WO 2013030750A1 IB 2012054381 W IB2012054381 W IB 2012054381W WO 2013030750 A1 WO2013030750 A1 WO 2013030750A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- imidazol
- amino
- methoxycarbonyl
- alkyl
- benzo
- Prior art date
Links
- 0 CC(*c(cc(CCc1ccc2nc(C3(C=CC=CC=C3)NC(OC(C)(C)C)=O)[n]c2c1)cc1)c1-c1c2nc(C(CCC3)N3C(OC(C)(C)C)=O)[n]1)C2=C Chemical compound CC(*c(cc(CCc1ccc2nc(C3(C=CC=CC=C3)NC(OC(C)(C)C)=O)[n]c2c1)cc1)c1-c1c2nc(C(CCC3)N3C(OC(C)(C)C)=O)[n]1)C2=C 0.000 description 13
- ITENZJVVWQPLEV-UMSFTDKQSA-N CC(C)(C)OC(NC1(CC1)c([nH]c1c2)nc1ccc2-c(cc1)ccc1-c1ccc(-c2c(CCC3)nc([C@H](CCC4)N4C(OC(C)(C)C)=O)[nH]2)c3c1)=O Chemical compound CC(C)(C)OC(NC1(CC1)c([nH]c1c2)nc1ccc2-c(cc1)ccc1-c1ccc(-c2c(CCC3)nc([C@H](CCC4)N4C(OC(C)(C)C)=O)[nH]2)c3c1)=O ITENZJVVWQPLEV-UMSFTDKQSA-N 0.000 description 1
- MVNNNHQVMDJHCI-DHUJRADRSA-N CC(C)(C)OC(NC1(CCC1)c1nc(ccc(-c(cc2)ccc2-c2ccc(-c3c(CCC4)nc([C@H](CCC5)N5C(OC(C)(C)C)=O)[nH]3)c4c2)c2)c2[nH]1)=O Chemical compound CC(C)(C)OC(NC1(CCC1)c1nc(ccc(-c(cc2)ccc2-c2ccc(-c3c(CCC4)nc([C@H](CCC5)N5C(OC(C)(C)C)=O)[nH]3)c4c2)c2)c2[nH]1)=O MVNNNHQVMDJHCI-DHUJRADRSA-N 0.000 description 1
- ANANGNZZYVLYSW-UHFFFAOYSA-N CC(C)(C)OC(NC1(CCCCC1)C(OC(CCc1cc(Br)ccc11)C1=O)=O)=O Chemical compound CC(C)(C)OC(NC1(CCCCC1)C(OC(CCc1cc(Br)ccc11)C1=O)=O)=O ANANGNZZYVLYSW-UHFFFAOYSA-N 0.000 description 1
- UKVJYHXUBFYJLP-UHFFFAOYSA-N CC(C)(C)OC(NC1(CCCCC1)c1nc(cc(B2OC(C)(C)C(C)(C)O2)cc2)c2[nH]1)=O Chemical compound CC(C)(C)OC(NC1(CCCCC1)c1nc(cc(B2OC(C)(C)C(C)(C)O2)cc2)c2[nH]1)=O UKVJYHXUBFYJLP-UHFFFAOYSA-N 0.000 description 1
- DBPIBCWSSOAYEQ-UHFFFAOYSA-N CC(C)(C)OC(NC1(CCCCC1)c1nc(cc(cc2)-c3ccc(B4OC(C)(C)C(C)(C)O4)cc3)c2[nH]1)=O Chemical compound CC(C)(C)OC(NC1(CCCCC1)c1nc(cc(cc2)-c3ccc(B4OC(C)(C)C(C)(C)O4)cc3)c2[nH]1)=O DBPIBCWSSOAYEQ-UHFFFAOYSA-N 0.000 description 1
- OTKLEPPYUCUXKZ-UHFFFAOYSA-N CC(C)(C)OC(NC1(CCCCC1)c1nc2cc(-c([s]3)ccc3Br)ccc2[nH]1)=O Chemical compound CC(C)(C)OC(NC1(CCCCC1)c1nc2cc(-c([s]3)ccc3Br)ccc2[nH]1)=O OTKLEPPYUCUXKZ-UHFFFAOYSA-N 0.000 description 1
- UFOOMNYITJSPKO-QXUSSCGESA-N CC(C)[C@@H](C(NC1(CCC1)c1nc(CCc2cc(-c(cc3)ccc3-c3ccc4nc([C@H](CCC5)N5C([C@H](C(C)C)NC(OC)=O)=O)[nH]c4c3)ccc2-2)c-2[nH]1)=O)NC(OC)=O Chemical compound CC(C)[C@@H](C(NC1(CCC1)c1nc(CCc2cc(-c(cc3)ccc3-c3ccc4nc([C@H](CCC5)N5C([C@H](C(C)C)NC(OC)=O)=O)[nH]c4c3)ccc2-2)c-2[nH]1)=O)NC(OC)=O UFOOMNYITJSPKO-QXUSSCGESA-N 0.000 description 1
- WHTOLBUGSNIXSB-PUBVIUOISA-N CC(C)[C@@H](C(NC1(CCC1)c1nc(ccc(-c2ccc(-c3cc4ccc5nc([C@H](CCC6)N6C([C@H](C(C)C)NC(OC)=O)=O)[nH]c5c4cc3)[s]2)c2)c2[nH]1)=O)NC(OC)=O Chemical compound CC(C)[C@@H](C(NC1(CCC1)c1nc(ccc(-c2ccc(-c3cc4ccc5nc([C@H](CCC6)N6C([C@H](C(C)C)NC(OC)=O)=O)[nH]c5c4cc3)[s]2)c2)c2[nH]1)=O)NC(OC)=O WHTOLBUGSNIXSB-PUBVIUOISA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
Definitions
- the present invention is related to novel compounds of the general formula I,
- HCV Persistent hepatitis C virus
- HCV chronic infection is often asymptomatic with latent periods lasting for decades before manifestation by which time extensive liver damage has occurred.
- HCV is spread primarily by unscreened blood transfusions and use of contaminated needles and syringes; the highest risk groups are intravenous drug users and people who received blood transfusions (mainly haemophiliacs) before 1990 when screening for HCV was introduced.
- Factors that have been reported to influence the rate of HCV disease progression include age (increasing age is associated with more rapid progression), gender (males have more rapid disease progression than females), alcohol consumption (associated with an increased rate of disease progression), HIV co-infection (associated with a markedly increased rate of disease progression), and fatty liver.
- HCV exists as many closely related viral sequences, termed as quasi-species, in the infected individual, making specific pharmaceutical targeting of HCV proteins challenging due to the rapid evolution of escape mutants. It is increasingly evident that a broad collection of specific, pan genotypic anti- viral drugs targeting multiple essential viral functions, in addition to the current viral therapies will be required for effective global control of HCV.
- HCV inhibitors Disclosures describing the synthesis of HCV inhibitors are US20090202478, US20090202483, WO2009020828, WO2009020825, WO2009102318, WO2009102325, WO2009102694, WO2008144380, WO2008021927, WO2008021928, WO2008021936, WO2006133326, WO2004014852, WO2008070447, WO2009034390, WO2006079833, WO2007031791 , WO2007070556, WO2007070600, WO2008064218, WO2008154601 , WO2007082554, WO2008048589, EP2121697, US8008264, US8008263, US201 10217265, US201 10217261 , US8012982, US8012942, US8012941 , US201 10223134, WO201 1 106992, WO201 1 106929, US201 10237636, US201
- the present invention provides a pharmaceutical composition, containing the compound of the general formula (I) as defined herein, its tautomeric forms, its stereoisomers, its analogs, its prodrugs, its isotopes, its N-oxides, its metabolites, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates, or its co-crystals in combination with the usual pharmaceutically employed carriers, diluents and the like are useful for the treatment of HCV infection.
- HCV is a member of the Flaviviridae family of enveloped, positive stranded RNA viruses belonging to the genus Hepacivirus.
- the genome is a single ⁇ 9.6kb strand of RNA and consists of one open reading frame that encodes for a polyprotein of -3000 amino acids flanked by untranslated regions at both 5' and 3' ends.
- This precursor polyprotein is then processed by viral and cellular proteases to yield 10 separate mature viral proteins critical for replication and assembly of progeny viral particles.
- the organization of the structural and non-structural proteins in the HCV polyprotein is as follows: C-E1 -E2-P7-NS2-NS3- NS4a-NS4b-NS5a-NS5b.
- NS2 is a zinc dependent metalloproteinase that functions in conjunction with a part of NS3 protein.
- NS3 protein has two catalytic activities associated with it: a serine protease at the N-terminal end which requires NS4A as a cofactor, and an ATPase dependent helicase activity at the C-terminal end.
- NS5A is a membrane anchored phosphoprotein that is present in basally phosphorylated (56kDa) and hyperphosphorylated (58kDa) forms.
- NS5A plays an important role in replication and infectivity of HCV.
- the NS5B protein encodes an RNA dependent RNA polymerase activity, key to the generation of progeny viruses. While the pathology of HCV infection mainly affects the liver, the virus is found in other cell types in the body including peripheral blood lymphocytes [Thomson BJ et al., Clin Microbial Infect. 2005, V ⁇ _, 86-94; Moriishi K et al., Antivir. Chem. Chemother. 2003, 14, 285-297].
- Characterization of the replicase machinery required for HCV RNA synthesis has defined the protease/helicase NS3 protein, the NS4A cofactor, the NS4B integral membrane protein, the NS5A protein and the RNA dependent RNA polymerase NS5B as being its essential components.
- R A is independently selected at each occurrence from-
- Ring 'A' is selected from 3 to 6 membered carbocycle and 3 to 6 membered heterocycle, the ring 'A' may further be annulated with substituted- or unsubstituted- carbocycle, substituted- or unsubstituted- heterocycle, substituted- or unsubstituted- aromatic carbocycle or substituted- or unsubstituted- aromatic heterocycle;
- Ring ⁇ ' is 5 to 10 membered heterocycle, the ring ⁇ ' may be monocyclic, fused bicyclic, bridged bicyclic or spiro bicyclic;
- R 3 is selected from O and N(R 13 );
- R 6 and R 7 are independently selected from the group consisting of hydrogen, halogen, substituted- or unsubstituted- Ci -6 alkyl, and R 10 O-;
- R 8 is selected from hydrogen and Ci -6 alkyl
- R 10 , R 11 , R 11a and R 12 are independently selected from hydrogen and substituted- or unsubstituted- Ci -6 alkyl;
- R 10a is substituted- or unsubstituted- Ci- 6 alkyl
- R 13 is selected from hydrogen and substituted- or unsubstituted- alkyl group
- R 14a is selected from hydrogen, alkyl, perhaloalkyl.
- any sub-range or individual number of carbon atoms falling within the indicated range also can be used.
- ring A is 3 to 6 membered carbocycle, such that at least once it is selected as
- R 3 is particularly selected as NH.
- R 4 is particularly selected from phenylene and five membered heteroarylene containing 1 or 2 heteroatoms selected from N and S, when n is particularly selected as 1 .
- R 4 is particularly selected as CR a R b , when n is particularly selected as 2 to form an ethylene or ethynylene linkage.
- n is particularly selected from 0, 1 and 2;
- R 5 , R 6 and R 7 are each particularly selected as hydrogen.
- Ring D is particularly selected from the group consisting of aromatic carbocycle, six membered carbocycle, seven membered carbocycle, and seven membered heterocycle containing one heteroatom particularly selected as oxygen.
- R A is selected independently at each occurrence from -
- g A is 3 to 6 membered carbocycle, such that at least once it is selected as
- R 3 is particularly selected as NH
- R 4 is particularly selected from the group consisting of CR a R b , phenylene and five membered heteroarylene containing 1 or 2 heteroatoms selected from N and S
- n is particularly selected from 0, 1 and 2
- R 5 , R 6 and R 7 are each particularly selected as hydrogen
- Ring D is particularly selected from the group consisting of aromatic carbocycle, six membered carbocycle, seven membered carbocycle, and seven membered heterocycle containing one heteroatom particularly selected as oxygen.
- alkyl means a straight chain or branched hydrocarbon containing from 1 to 20 carbon atoms. Preferably the alkyl chain may contain 1 to 10 carbon atoms. More preferably alkyl chain may contain up to 6 carbon atoms. Representative examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, and n- hexyl.
- haloalkyl used herein means an alkyl group as defined hereinabove wherein at least one of the hydrogen atoms of the said alkyl group is substituted with halogen.
- the haloalkyl group is exemplified by monofluoromethyl, 1 ,2-dichloroethyl and the like.
- perhaloalkyl means an alkyl group as defined hereinabove wherein all the hydrogen atoms of the said alkyl group are substituted with halogen.
- the perhaloalkyl group is exemplified by trifluoromethyl, pentafluoroethyl and the like.
- cycloalkyi means a monocyclic, bicyclic, or tricyclic non- aromatic ring system containing from 3 to 14 carbon atoms, preferably monocyclic cycloalkyi ring containing 3 to 6 carbon atoms.
- monocyclic ring systems include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- Bicyclic ring systems are also exemplified by a bridged monocyclic ring system in which two non-adjacent carbon atoms of the monocyclic ring are linked by an alkylene bridge.
- bicyclic ring systems include, but are not limited to, bicyclo[3.1 .1 ]heptane, bicyclo[2.2.1 ]heptane, bicyclo[2.2.2]octane, bicyclo[3.2.2]nonane, bicyclo[3.3.1 ]nonane, and bicyclo[4.2.1 ]nonane, bicyclo[3.3.2]decane, bicyclo[3.1 .0]hexane, bicyclo[410]heptane, bicyclo[3.2.0]heptanes, octahydro-1 H-indene.
- Tricyclic ring systems are also exemplified by a bicyclic ring system in which two non- adjacent carbon atoms of the bicyclic ring are linked by a bond or an alkylene bridge.
- Representative examples of tricyclic-ring systems include, but are not limited to, tricyclo[3.3.1 .0 3 7 ]nonane and tricyclo[3.3.1 .1 3 7 ]decane (adamantane).
- cycloalkyi also include spiro systems wherein one of the ring is annulated on a single carbon atom such ring systems are exemplified by spiro[2.5]octane, spiro[4.5]decane, spiro[bicyclo[4.1 .0]heptane-2,1 '-cyclopentane], hexahydro-2'H-spiro[cyclopropane-1 , 1 '- pentalene].
- cycloalkenyl as used herein, means a cycloalkyi group as defined above containing at least one double bond.
- Carbocycle as used herein, means a cyclic system made up of carbon atoms, which includes cycloalkane, cycloalkene and aromatic carbocycle.
- aryl refers to a monovalent monocyclic, bicyclic or tricyclic aromatic hydrocarbon ring system.
- aryl groups include phenyl, naphthyl, anthracenyl, fluorenyl, indenyl, azulenyl, and the like.
- Aryl group also include partially saturated bicyclic and tricyclic aromatic hydrocarbons such as tetrahydro-naphthalene.
- the said aryl group also includes aryl rings fused with heteroaryl or heterocyclic rings such as 2,3-dihydro- benzo[1 ,4]dioxin-6-yl, 2,3-dihydro-benzo[1 ,4]dioxin-5-yl, 2,3-dihydro-benzofuran-5-yl, 2,3- dihydro-benzofuran-4-yl, 2,3-dihydro-benzofuran-6-yl, 2,3-dihydro-benzofuran-6-yl, 2,3- dihydro-1 H-indol-5-yl, 2,3-dihydro-1 H-indol-4-yl, 2,3-dihydro-1 H-indol-6-yl, 2,3-dihydro-1 H- indol-7-yl, benzo[1 ,3]dioxol-4-yl, benzo[1 ,3]dioxol-5-yl
- substituents selected independently from the group consisting of halogen, nitro, cyano, hydroxy, Ci to C 6 al
- heteroaryl refers to a 5- 14 membered monocyclic, bicyclic, or tricyclic ring system having 1 -4 ring heteroatoms selected from O, N, or S, and the remainder ring atoms being carbon (with appropriate hydrogen atoms unless otherwise indicated), wherein at least one ring in the ring system is aromatic. Heteroaryl groups may be optionally substituted with one or more substituents. In one embodiment, 0, 1 , 2, 3, or 4 atoms of each ring of a heteroaryl group may be substituted by a substituent.
- heteroaryl groups include pyridyi, 1 -oxo-pyridyl, furany!, thienyl, pyrroiyL oxazolyl, oxadiazolyl, imidazolyl, thiazolyl, isoxazolyl, quinolinyl, pyrazolyl, isothiazolyl, pyridazinyL pyrimidinyl, pyrazinyl, triazinyl. triazolyl.
- thiadiazolyl isoquinolinyl, benzoxazolyl, benzofuranyl, indolizinyl, imidazopyridyl, tetrazolyl, benzimidazolyi, benzothiazolyl, benzothiadiazolyl, benzoxadiazolyl, indolyl, azaindolyL imidazopyridyl, quinazolinyl, purinyl, pyrrolo[2,3]pyrimidinyi, pyrazolo[3,4]pyrimidinyl, and benzo(b)thienyi, 2,3- thiadiazolyL 1 H-pyrazoio[5,1 -c]-1 ,2,4-triazoiyi, pyrro!o[3,4-d]-1 ,2,3-triazolyi, cyclopentatriazolyl, 3H-pyrrolo[3,4-c] isoxazoly! and the like.
- substituents selected independently from the group consisting of halogen, nitro, cyano, hydroxy, Ci
- heterocycle or “heterocyclic” as used herein, means a 'cycloalkyi' group wherein one or more of the carbon atoms replaced by -0-, -S-, -S(0 2 )-, -S(O)-, -N(R m )-, - Si(R m )R n -, wherein, R m and R n are independently selected from hydrogen, alkyl, aryl, heteroaryl, cycloalkyi, and heterocyclyl.
- the heterocycle may be connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the heterocycle.
- monocyclic heterocycle include, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1 ,3-dioxanyl, 1 ,3-dioxolanyl, 1 ,3- dithiolanyl, 1 ,3-dithianyl, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl.
- oxadiazolidinyl oxazolinyl, oxazolidinyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl.
- thiopyranyl, and trithianyl thiopyranyl, and trithianyl.
- bicyclic heterocycle include, but are not limited to 1 ,3-benzodioxolyl, 1 ,3-benzodithiolyl, 2,3-dihydro-1 ,4-benzodioxinyl, 2,3- dihydro-1 -benzofuranyl, 2,3-dihydro-1 -benzothienyl, 2,3-dihydro-1 H-indolyl and 1 ,2,3,4- tetrahydroquinolinyl.
- the term heterocycle also include bridged heterocyclic systems such as azabicyclo[3.2.1 ]octane, azabicyclo[3.3.1 ]nonane and the like.
- oxo attached to carbon forms a carbonyl
- oxo substituted on cyclohexane forms a cyclohexanone
- 'annulated' means the ring system under consideration is either annulated with another ring at a carbon atom of the cyclic system or across a bond of the cyclic system as in the case of fused or spiro ring systems.
- bridged' means the ring system under consideration contain an alkylene bridge having 1 to 4 methylene units joining two non-adjacent ring atoms.
- the compounds of general formula I where all the symbols are as defined earlier can be prepared by methods given in Schemes given below or in the examples; the disclosure should not be construed to limit the scope of the invention arriving at compound of formula I as disclosed hereinabove.
- the benzimidazole derivatives 4 could be prepared by coupling different ring sized cyclic (carbocyclic or heterocyclic) amino acids 2 with 1 ,2-diamino-4-bromobenzene 1 .
- the coupling of 1 with Boc-protected amino acid 2 is carried out either by using conventional coupling reagents such as EDCI or HATU could provide a mixture of amide derivatives 3, Scheme 1 .
- 3 could be prepared by the conversion of the protected amino acids to acid chloride by isobutyl chloroformate in the presence of a base and subsequent addition to 1 ,2-diamino-4-bromobenzene in the presence of an organic base such as Et 3 N or N-methylmorpholine.
- the mixture of regio isomers can then be cyclised to the benzimidazole derivative 4 in presence of acetic acid or P 2 0 5 or by simple reflux with ethanol for extended periods.
- the tricyclic units of different ring size 6 can be synthesized by a number of possible methods from the easily accessible tetralone moiety 7 as provided in WO2009102633.
- alpha-bromo ketone 8 synthesized by addition of molecular bromine to the tetralone moiety 7, on O-Alkylation with an amino acid such as N-Boc proline could provide intermediate 9 which can be cyclised to the imidazole containing tricycle 6 using NH 4 OAc.
- the oximino-ketone 10, prepared by the addition of alkyl nitrite to the tetralone moiety 7, can be cyclised with an aldehyde such as N-Boc-Prolinal in the presence of ammonium acetate to the hydroxylated imidazole derivative 11 using the process provided in Bioorg. Med. Chem. Lett., 2002, 1009-101 1 .
- Treatment of 11 with triethyl phosphite can provide the imidazole containing tricycle 6.
- Another approach could be from the alpha-amino ketone 13, which can be synthesized from the oximino-ketone 10 or the tetralone moiety 7, Scheme 2.
- Amide coupling with an N-Boc protected amino acid such as Proline could provide the amide 14 which can be cyclised to the imidazole containing tricycle 6 in the presence of ammonium acetate.
- X 2 ,Y 2 . Z 2 are independently selected from C and N
- the alkyne linker can be reduced to ethylene linker by catalytic hydrogenation to give compounds of formula 31 , Scheme 7, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
- the aldehyde can be reduced to an alcohol and the alcohol can be transformed to a suitable leaving group such as tosylate, mesylate, bromide, etc to give intermediate 35, which can be reacted with phenolic benzimidazoles 36 to give compound 37, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
- a suitable leaving group such as tosylate, mesylate, bromide, etc.
- R 4 any cyclic system
- R 4 -0-, -OCH 2 -, -(CH 2 ) 2 , etc.
- L is a leaving group
- R 1 , R 3 , R 6 , R 7 , R 8 , R 9 , m, n, rings A and D are as defined under compound of formula I
- Scheme 1 1 shows final step in the synthesis of compound of formula I.
- Ring substitutions viz. R 1 , R 2 , R 6 and R 7 may be incorporated or transformed from one to another by known functional group transformation techniques at any of the steps provided hereinabove during synthesis of compound of formula I (schemes 1 to 1 1 ) or they may be protected/de-protected using suitable protecting groups wherever required using the known protection de-protection techniques as provided in Greene and Wuts 'protective groups in Organic Synthesis', Wiley and sons, 1999.
- the intermediates and the compounds of the present invention are obtained in pure form in a manner known per se, for example by distilling off the solvent in vacuum and re- crystallizing the residue obtained from a suitable solvent, such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations or subjecting it to one of the purification methods, such as column chromatography (eg. flash chromatography) on a suitable support material such as alumina or silica gel using eluent such as dichloromethane, ethyl acetate, hexane, methanol, acetone and their combinations.
- a suitable solvent such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations
- the purification methods such as column chromatography (eg. flash chromatography) on
- Salts of compound of formula I are obtained by dissolving the compound in a suitable solvent, for example in a chlorinated hydrocarbon, such as methyl chloride or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol, which was then treated with the desired acid or base as described in Berge S.M. et al. "Pharmaceutical Salts, a review article in Journal of Pharmaceutical sciences volume 66, page 1 -19 (1977)" and in handbook of pharmaceutical salts properties, selection, and use by P.H.Einrich Stahland Camille G.wermuth , Wiley- VCH (2002).
- a suitable solvent for example in a chlorinated hydrocarbon, such as methyl chloride or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol
- stereoisomers of the compounds of formula I, or intermediates thereof in the present invention may be prepared by stereospecific syntheses or resolution of the racemic compound using an optically active amine, acid or complex forming agent, and separating the diastereomeric salt/complex by fractional crystallization or by column chromatography.
- Compounds of the present invention were prepared using synthetic schemes provided below:
- a further embodiment of the present invention includes pharmaceutical compositions comprising any single compound, a combination of two or more compounds delineated herein, or a pharmaceutically acceptable salt thereof, with a pharmaceutically acceptable carrier or excipient.
- a further embodiment of the present invention is a pharmaceutical composition
- a pharmaceutical composition comprising any single compound or a combination of two or more compounds delineated herein, or a pharmaceutically acceptable salt thereof, in combination with one or more agents known in the art, with a pharmaceutically acceptable carrier or excipient.
- compounds of the present invention can be administered as the sole active pharmaceutical agent, or used in combination with one or more agents to treat or prevent hepatitis C infections or the symptoms associated with HCV infection.
- Other agents to be administered in combination with a compound or combination of compounds of the present invention include therapies for diseases caused by HCV infection that suppresses HCV viral replication by direct or indirect mechanisms.
- agents include, but not limited to, host immune modulators (for example, interferon-alpha, pegylated interferon-alpha, consensus interferon, interferon-beta, interferon-gamma, CpG oligonucleotides and the like); antiviral compounds that inhibit host cellular functions such as inosine monophosphate dehydrogenase (for example, ribavirin and the like); cytokines that modulate immune function (for example, interleukin 2, interleukin 6, and interleukin 12); a compound that enhances the development of type 1 helper T cell response; interfering RNA; anti-sense RNA; vaccines comprising HCV antigens or antigen adjuvant combinations directed against HCV; agents that interact with host cellular components to block viral protein synthesis by inhibiting the internal ribosome entry site (IRES) initiated translation step of HCV viral replication or to block viral particle maturation and release with agents targeted toward the viroporin family of membrane proteins such as, for example
- compositions of the present invention may further comprise inhibitor(s) of other targets in the HCV life cycle, including, but not limited to, helicase, polymerase, metalloprotease, NS4A protein, NS5A protein, and internal ribosome entry site (IRES).
- inhibitor(s) of other targets in the HCV life cycle including, but not limited to, helicase, polymerase, metalloprotease, NS4A protein, NS5A protein, and internal ribosome entry site (IRES).
- one embodiment of the present invention is directed to a method for treating or preventing an infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment one or more agents selected from the group consisting of a host immune modulator and a second or more antiviral agents, or a combination thereof, with a therapeutically effective amount of a compound or combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof.
- Examples of the host immune modulator are, but not limited to, interferon-alpha, pegylated-interferon-alpha, interferon-beta, interferon-gamma, a cytokine, a vaccine, and a vaccine comprising an antigen and an adjuvant, and said second antiviral agent inhibits replication of HCV either by inhibiting host cellular functions associated with viral replication or by targeting proteins of the viral genome.
- Example of the RNA-containing virus includes, but not limited to, hepatitis C virus (HCV).
- a further embodiment of the present invention is directed to a method of treating or preventing infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment an agent or combination of agents that treat or alleviate symptoms of HCV infection including cirrhosis and inflammation of the liver, with a therapeutically effective amount of a compound or combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof.
- RNA-containing virus includes, but not limited to, hepatitis C virus (HCV).
- Yet another embodiment of the present invention provides a method of treating or preventing infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment one or more agents that treat patients for disease caused by hepatitis B (HBV) infection, with a therapeutically effective amount of a compound or a combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof.
- An agent that treats patients for disease caused by hepatitis B (HBV) infection may be for example, but not limited thereto, L-deoxythymidine, adefovir, lamivudine or tenfovir, or any combination thereof.
- Example of the RNA-containing virus includes, but not limited to, hepatitis C virus (HCV).
- a further embodiment of the present invention provides a method of treating or preventing infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment one or more agents that treat patients for disease caused by human immunodeficiency virus (HIV) infection, with a therapeutically effective amount of a compound or a combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof.
- HIV human immunodeficiency virus
- the agent that treats patients for disease caused by human immunodeficiency virus (H IV) infection may include, but is not limited thereto, ritonavir, lopinavir, indinavir, nelfmavir, saquinavir, amprenavir, atazanavir, tipranavir, TMC-1 14, fosamprenavir, zidovudine, lamivudine, didanosine, stavudine, tenofovir, zalcitabine, abacavir, efavirenz, nevirapine, delavirdine, TMC-125, L-870812, S-1360, enfuvirtide (T- 20) or T-1249, or any combination thereof.
- RNA-containing virus includes, but not limited to, hepatitis C virus (HCV). It can occur that a patient may be co-infected with hepatitis C virus and one or more other viruses, including but not limited to human immunodeficiency virus (HIV), hepatitis A virus (HAV) and hepatitis B virus (HBV). Thus also contemplated is combination therapy to treat such co-infections by co-administering a compound according to the present invention with at least one of an HIV inhibitor, an HAV inhibitor and an HBV inhibitor.
- HCV human immunodeficiency virus
- HAV hepatitis A virus
- HBV hepatitis B virus
- the present invention provides the use of a compound or a combination of compounds of the invention, or a therapeutically acceptable salt thereof, and one or more agents selected from the group consisting of a host immune modulator and a second or more antiviral agents, or a combination thereof, to prepare a medicament for the treatment of an infection caused by an RNA-containing virus in a patient, particularly hepatitis C virus.
- host immune modulators include, but are not limited to, interferon- alpha, pegylated-interferon-alpha, interferon-beta, interferon-gamma, a cytokine, a vaccine, and a vaccine comprising an antigen and an adjuvant, and said second antiviral agent inhibits replication of HCV either by inhibiting host cellular functions associated with viral replication or by targeting proteins of the viral genome.
- combination of compound or compounds of the present invention, together with one or more agents as defined herein above can be employed in pure form or, where such forms exist, in pharmaceutically acceptable salt thereof.
- combination of therapeutic agents can be administered as a pharmaceutical composition containing a therapeutically effective amount of the compound or combination of compounds of interest, or their pharmaceutically acceptable salt thereof, in combination with one or more agents as defined hereinabove, and a pharmaceutically acceptable carrier.
- Such pharmaceutical compositions can be used for inhibiting the replication of an RNA-containing virus, particularly Hepatitis C virus (HCV), by contacting said virus with said pharmaceutical composition.
- such compositions are useful for the treatment or prevention of an infection caused by an RNA-containing virus, particularly Hepatitis C virus (HCV).
- a still further embodiment of the invention is directed to a method of treating or preventing infection caused by an RNA-containing virus, particularly a hepatitis C virus (HCV), comprising administering to a patient in need of such treatment a pharmaceutical composition comprising a compound or combination of compounds of the invention or a pharmaceutically acceptable salt thereof, and one or more agents as defined hereinabove, with a pharmaceutically acceptable carrier.
- an RNA-containing virus particularly a hepatitis C virus (HCV)
- HCV hepatitis C virus
- the therapeutic agents When administered as a combination, the therapeutic agents can be formulated as separate compositions which are given at the same time or within a predetermined period of time, or the therapeutic agents can be given as a single unit dosage form.
- Antiviral agents contemplated for use in such combination therapy include agents (compounds or biologicals) that are effective to inhibit the formation and/or replication of a virus in a mammal, including but not limited to agents that interfere with either host or viral mechanisms necessary for the formation and/or replication of a virus in a mammal.
- agents can be selected from another anti-HCV agent; an HIV inhibitor; an HAV inhibitor; and an HBV inhibitor.
- agents to be administered in combination with a compound of the present invention include a cytochrome P450 monooxygenase inhibitor, which is expected to inhibit metabolism of the compounds of the invention. Therefore, the cytochrome P450 monooxygenase inhibitor would be in an amount effective to inhibit metabolism of the compounds of the present invention. Accordingly, the CYP inhibitor is administered in an amount such that the bioavailability of the compounds of the present invention is increased in comparison to the bioavailability in the absence of the CYP inhibitor.
- room temperature used in the specification denotes any temperature ranging between about 20 °C to about 40 °C, except and otherwise it is specifically mentioned in the specification.
- the intermediates and the compounds of the present invention may obtained in pure form in a manner known per se, for example, by distilling off the solvent in vacuum and re- crystallizing the residue obtained from a suitable solvent, such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations or subjecting it to one of the purification methods, such as column chromatography (e.g., flash chromatography) on a suitable support material such as alumina or silica gel using eluent such as dichloromethane, ethyl acetate, hexane, methanol, acetone and their combinations.
- a suitable solvent such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations
- the purification methods such as column chromatography (e.g., flash
- Salts of compound of formula I can be obtained by dissolving the compound in a suitable solvent, for example in a chlorinated hydrocarbon, such as methyl chloride or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol, which was then treated with the desired acid or base as described in Berge S.M. et al. "Pharmaceutical Salts, a review article in Journal of Pharmaceutical sciences volume 66, page 1 -19 (1977)" and in handbook of pharmaceutical salts properties, selection, and use by P.H.Einrich Stahland Camille G.wermuth, Wiley- VCH (2002).
- a suitable solvent for example in a chlorinated hydrocarbon, such as methyl chloride or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol
- salts can also be found in Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p. 1445, and Journal of Pharmaceutical Science, 66, 2-19 (1977).
- they can be a salt of an alkali metal (e.g., sodium or potassium), alkaline earth metal (e.g., calcium), or ammonium of salt.
- the compound of the invention or a composition thereof can potentially be administered as a pharmaceutically acceptable acid-addition, base neutralized or addition salt, formed by reaction with inorganic acids, such as hydrochloric acid, hydrobromic acid, perchloric acid, nitric acid, thiocyanic acid, sulfuric acid, and phosphoric acid, and organic acids such as formic acid, acetic acid, propionic acid, glycolic acid, lactic acid, pyruvic acid, oxalic acid, malonic acid, succinic acid, maleic acid, and fumaric acid, or by reaction with an inorganic base, such as sodium hydroxide, potassium hydroxide.
- inorganic acids such as hydrochloric acid, hydrobromic acid, perchloric acid, nitric acid, thiocyanic acid, sulfuric acid, and phosphoric acid
- organic acids such as formic acid, acetic acid, propionic acid, glycolic acid, lactic acid, pyruvic acid, oxalic acid, mal
- the conversion to a salt is accomplished by treatment of the base compound with at least a stoichiometric amount of an appropriate acid.
- the free base is dissolved in an inert organic solvent such as diethyl ether, ethyl acetate, chloroform, ethanol, methanol, and the like, and the acid is added in a similar solvent.
- the mixture is maintained at a suitable temperature (e.g., between 0 °C and 50 °C).
- the resulting salt precipitates spontaneously or can be brought out of solution with a less polar solvent.
- the stereoisomers of the compounds of formula I of the present invention may be prepared by stereospecific syntheses or resolution of the achiral compound using an optically active amine, acid or complex forming agent, and separating the diastereomeric salt/complex by fractional crystallization or by column chromatography.
- prodrug denotes a derivative of a compound, which derivative, when administered to warm-blooded animals, e.g. humans, is converted into the compound (drug).
- the enzymatic and/or chemical hydrolytic cleavage of the compounds of the present invention occurs in such a manner that the proven drug form (parent carboxylic acid drug) is released, and the moiety or moieties split off remain nontoxic or are metabolized so that nontoxic metabolic products are produced.
- a carboxylic acid group can be esterified, e.g., with a methyl group or ethyl group to yield an ester.
- an ester is administered to a subject, the ester is cleaved, enzymatically or non- enzymatically, reductively, oxidatively, or hydrolytically, to reveal the anionic group.
- An anionic group can be esterified with moieties (e.g., acyloxymethyl esters) which are cleaved to reveal an intermediate compound which subsequently decomposes to yield the active compound.
- the prodrugs can be prepared in situ during the isolation and purification of the compounds, or by separately reacting the purified compound with a suitable derivatizing agent.
- hydroxy groups can be converted into esters via treatment with a carboxylic acid in the presence of a catalyst.
- cleavable alcohol prodrug moieties include substituted or unsubstituted, branched or unbranched lower alkyl ester moieties, e.g., ethyl esters, di-lower alkylamino lower-alkyl esters, e.g., dimethylaminoethyl ester, acylamino lower alkyl esters, acyloxy lower alkyl esters (e.g., pivaloyloxymethyl ester), aryl esters, e.g., phenyl ester, aryl-lower alkyl esters, e.g., benzyl ester, optionally substituted, e.g., with methyl, halo, or methoxy substituents aryl and aryl- lower alkyl esters, amides, lower-alkyl amides, di-lwer alkyl amides, and hydroxy amides.
- lower alkyl ester moieties
- the present invention further provides a pharmaceutical composition, containing the compounds of the general formula (I) as defined above, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates and its co-crystals in combination with the usual pharmaceutically acceptable carriers, diluents and the like.
- the pharmaceutically acceptable carrier is preferably one that is chemically inert to the compound of the invention and one that has no detrimental side effects or toxicity under the conditions of use.
- Such pharmaceutically acceptable carriers preferably include saline (e.g., 0.9% saline), Cremophor EL (which is a derivative of castor oil and ethylene oxide available from Sigma Chemical Co., St.
- a preferred pharmaceutical carrier is polyethylene glycol, such as PEG 400, and particularly a composition comprising 40% PEG 400 and 60% water or saline. The choice of carrier will be determined in part by the particular compound chosen, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of the pharmaceutical composition of the present invention.
- formulations for oral, aerosol, parenteral, subcutaneous, intravenous, intraarterial, intramuscular, interperitoneal, rectal, and vaginal administration are merely exemplary and are in no way limiting.
- compositions for parenteral administration that comprise a solution of the compound of the invention dissolved or suspended in an acceptable carrier suitable for parenteral administration, including aqueous and non-aqueous, isotonic sterile injection solutions.
- compositions include solutions containing anti-oxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives.
- the compound can be administered in a physiologically acceptable diluent in a pharmaceutical carrier, such as a sterile liquid or mixture of liquids, including water, saline, aqueous dextrose and related sugar solutions, an alcohol, such as ethanol, isopropanol (for example in topical applications), or hexadecyl alcohol, glycols, such as propylene glycol or polyethylene glycol, dimethylsulfoxide, glycerol ketals, such as 2,2-dimethyl-1 ,3-dioxolane- 4-methanol, ethers, such as poly(ethyleneglycol) 400, an oil, a fatty acid, a fatty acid ester or glyceride, or an acetylated fatty acid glyceride with or without the addition of a pharmaceutically acceptable surfactant, such as a soap or a detergent, suspending agent, such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carb
- Oils useful in parenteral formulations include petroleum, animal, vegetable, and synthetic oils. Specific examples of oils useful in such formulations include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral oil. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
- Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts
- suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylene polypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl-3-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
- the parenteral formulations typically will contain from about 0.5% or less to about 25% or more by weight of a compound of the invention in solution. Preservatives and buffers can be used. In order to minimize or eliminate irritation at the site of injection, such compositions can contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17. The quantity of surfactant in such formulations will typically range from about 5% to about 15% by weight. Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol.
- HLB hydrophile-lipophile balance
- parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampoules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use.
- sterile liquid excipient for example, water
- Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets.
- Formulations suitable for oral administration can consist of (a) liquid solutions, such as an effective amount of a compound of the invention dissolved in diluents, such as water, saline, or orange juice; (b) capsules, sachets, tablets, lozenges, and troches, each containing a pre-determined amount of the compound of the invention, as solids or granules; (c) powders; (d) suspensions in an appropriate liquid; and (e) suitable emulsions.
- Liquid formulations can include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent.
- diluents such as water and alcohols, for example, ethanol, benzyl alcohol, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent.
- Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and cornstarch.
- Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible excipients.
- Lozenge forms can comprise the compound ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising a compound of the invention in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the compound of the invention, such excipients as are known in the art.
- a flavor usually sucrose and acacia or tragacanth
- pastilles comprising a compound of the invention in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the compound of the invention, such excipients as are known in the art.
- a compound of the present invention can be made into aerosol formulations to be administered via inhalation.
- a compound or epimer of the invention is preferably supplied in finely divided form along with a surfactant and propellant.
- Typical percentages of the compounds of the invention can be about 0.01 % to about 20% by weight, preferably about 1 % to about 10% by weight.
- the surfactant must, of course, be nontoxic, and preferably soluble in the propellant.
- Such surfactants are the esters or partial esters of fatty acids containing from 6 to 22 carbon atoms, such as caproic, octanoic, lauric, palmitic, stearic, linoleic, linolenic, olesteric and oleic acids with an aliphatic polyhydric alcohol or its cyclic anhydride.
- Mixed esters such as mixed or natural glycerides can be employed.
- the surfactant can constitute from about 0.1 % to about 20% by weight of the composition, preferably from about 0.25% to about 5%.
- the balance of the composition is ordinarily propellant.
- a carrier can also be included as desired, e.g., lecithin, for intranasal delivery.
- aerosol formulations can be placed into acceptable pressurized propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like. They also can be formulated as pharmaceuticals for non-pressured preparations, such as in a nebulizer or an atomizer. Such spray formulations can be used to spray mucosa.
- acceptable pressurized propellants such as dichlorodifluoromethane, propane, nitrogen, and the like.
- non-pressured preparations such as in a nebulizer or an atomizer.
- Such spray formulations can be used to spray mucosa.
- the compound of the invention can be made into suppositories by mixing with a variety of bases, such as emulsifying bases or water-soluble bases.
- bases such as emulsifying bases or water-soluble bases.
- Formulations suitable for vaginal administration can be presented as pessaries, tampons, creams, gels, pastes, foams, or spray formulas containing, in addition to the compound ingredient, such carriers as are known in the art to be appropriate.
- the concentration of the compound in the pharmaceutical formulations can vary, e.g., from less than about 1 % to about 10%, to as much as 20% to 50% or more by weight, and can be selected primarily by fluid volumes, and viscosities, in accordance with the particular mode of administration selected.
- a typical pharmaceutical composition for intravenous infusion could be made up to contain 250 ml of sterile Ringer's solution, and 100 mg of at least one compound of the invention.
- Actual methods for preparing parenterally administrable compounds of the invention will be known or apparent to those skilled in the art and are described in more detail in, for example, Remington's Pharmaceutical Science (17 th ed., Mack Publishing Company, Easton, PA, 1985).
- the compound of the invention can be formulated as inclusion complexes, such as cyclodextrin inclusion complexes, or liposomes.
- Liposomes can serve to target a compound of the invention to a particular tissue, such as lymphoid tissue or cancerous hepatic cells. Liposomes can also be used to increase the half-life of a compound of the invention. Many methods are available for preparing liposomes, as described in, for example, Szoka et al., Ann. Rev. Biophys. Bioeng., 9, 467 (1980) and U.S. Patents 4,235,871 , 4,501 ,728, 4,837,028, and 5,019,369.
- the present invention also provides a pharmaceutical composition, containing the compounds of the general formula (I) as defined above, its tautomeric forms, its stereoisomers, its analogs, its prodrugs, its isotopes, its metabolites, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates and its co- crystals in combination with the usual pharmaceutically employed carriers, diluents and the like, and for use in any of the methods described herein.
- the compounds of the invention can be administered in a dose sufficient to treat the disease, condition or disorder.
- doses are known in the art (see, for example, the Physicians' Desk Reference (2004)).
- the compounds can be administered using techniques such as those described in, for example, Wasserman et al., Cancer, 36, pp. 1258-1268 (1975) and Physicians' Desk Reference, 58th ed., Thomson PDR (2004).
- Suitable doses and dosage regimens can be determined by conventional range-finding techniques known to those of ordinary skill in the art. Generally, treatment is initiated with smaller dosages that are less than the optimum dose of the compound of the present invention. Thereafter, the dosage is increased by small increments until the optimum effect under the circumstances is reached.
- the present method can involve the administration of about 0.1 ⁇ g to about 50 mg of at least one compound of the invention per kg body weight of the individual.
- dosages of from about 10 ⁇ g to about 200 mg of the compound of the invention would be more commonly used, depending on a patient's physiological response.
- the dose of the pharmaceutically active agent(s) described herein for methods of treating or preventing a disease or condition as described above can be about 0.001 to about 1 mg/kg body weight of the subject per day, for example, about 0.001 mg, 0.002 mg, 0.005 mg, 0.010 mg, 0.015 mg, 0.020 mg, 0.025 mg, 0.050 mg, 0.075 mg, 0.1 mg, 0.15 mg, 0.2 mg, 0.25 mg, 0.5 mg, 0.75 mg, or 1 mg/kg body weight per day.
- the dose of the pharmaceutically active agent(s) described herein for the described methods can be about 1 to about 1000 mg/kg body weight of the subject being treated per day, for example, about 1 mg, 2 mg, 5 mg, 10 mg, 15 mg, 0.020 mg, 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 500 mg, 750 mg, or 1000 mg/kg body weight per day.
- the present invention provides methods of treating, preventing, ameliorating, and/or inhibiting a hepatitis C virus infection comprising administering a compound of formula (I) or a salt thereof.
- the compounds of the present invention are effective against the HCV 1 b genotype. It should also be understood that the compounds of the present invention can inhibit multiple genotypes of HCV. Hence, in one of the embodiment compound of the present invention are active against the 1 a, 1 b, 2a, 2b, 3a, 4a, and 5a genotypes.
- inventive methods can provide any amount of any level of treatment, prevention, amelioration, or inhibition of the disorder in a mammal.
- a disorder, including symptoms or conditions thereof may be reduced by, for example, 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, or 10%.
- the treatment, prevention, amelioration, or inhibition provided by the inventive method can include treatment, prevention, amelioration, or inhibition of one or more conditions or symptoms of the disorder, e.g., cancer.
- treatment,” “prevention,” “amelioration,” or “inhibition” can encompass delaying the onset of the disorder, or a symptom or condition thereof.
- the term subject includes an "animal" which in turn includes a mammal such as, without limitation, the order Rodentia, such as mice, and the order Lagomorpha, such as rabbits.
- a mammal such as, without limitation, the order Rodentia, such as mice, and the order Lagomorpha, such as rabbits.
- the mammals are from the order Carnivora, including Felines (cats) and Canines (dogs). It is more preferred that the mammals are from the order Artiodactyla, including Bovines (cows) and Swine (pigs) or of the order Perssodactyla, including Equines (horses). It is most preferred that the mammals are of the order Primates, Ceboids, or Simoids (monkeys) or of the order Anthropoids (humans and apes). An especially preferred mammal is the human.
- viral infection refers to the introduction of a virus into cells or tissues, e.g., hepatitis C virus (HCV). In general, the introduction of a virus is also associated with replication. Viral infection may be determined by measuring virus antibody titer in samples of a biological fluid, such as blood, using, e.g., enzyme immunoassay. Other suitable diagnostic methods include molecular based techniques, such as RT-PCR, direct hybrid capture assay, nucleic acid sequence based amplification, and the like. A virus may infect an organ, e.g., liver, and cause disease, e.g., hepatitis, cirrhosis, chronic liver disease and hepatocellular carcinoma.
- HCV hepatitis C virus
- immune modulator refers to any substance meant to alter the working of the humoral or cellular immune system of a subject.
- immune modulators include inhibitors of mast cell-mediated inflammation, interferons, interleukins, prostaglandins, steroids, cortico-steroids, colony-stimulating factors, chemotactic factors, etc.
- ACN Acetonitrile. Boc: fert-butyloxycarbonyl.
- CDCI 3 Deuterochloroform.
- DCM dichloromethane.
- DDQ 2,3-Dichloro-5,6-dicyano-p-benzoquinone.
- DIPEA N, N- diisopropylethylamine.
- DME 1 , 2-dimethoxyethane.
- DMF N, N-dimethylformamide.
- DMSO-c 6 Dimethyl sulfoxide-d 6 .
- EDCI A/-(3-Dimethylaminopropyl)-A/'-ethylcarbodiimide.
- EtOAc Ethyl acetate.
- HATU 0-(7-Azabenzotriazol-1 -yl)-A/,/ ⁇ /,/ ⁇ /',/ ⁇ /'-tetramethyluronium hexafluorophosphate.
- HCI hydrochloric acid.
- HOBt 1 -Hydroxybenzotriazole hydrate.
- CD 3 OD Methanol-d 4 .
- Na 2 S0 4 sodium sulphate.
- NaHC0 3 sodium bicarbonate.
- Pd(PPh 3 ) 4 Pd(PPh 3 ) 4 :
- the vial was sealed and irradiated under microwave at 1 15 °C for 45 min. After cooling, the reaction mixture was added to water and the reaction mass was extracted with 20 % CHCI 3 -MeOH and dried over Na 2 S0 4 and concentrated. Purification was done using CombiflashTM by eluting with 2-5 % MeOH in DCM to yield the title compound as a white foam (300 mg, 68%).
- Step 1 tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (1 a)
- the mixture of isomers as synthesized above was taken in 10 mL of acetic acid and heated at 60 °C for 4 h. The reaction was cooled and most of the acetic acid was removed in the rotavap and the remaining traces of acid was neutralised with a satd. solution of NaHC0 3 . The compound was extracted with 100 mL ethyl acetate twice. The combined ethyl acetate extracts were dried over Na 2 S0 4 and concentrated in the rotavap to obtain a brown mass which was purified by column chromatography (silica gel; 20-30% EtOAc/hexane) to yield a pale brown coloured solid (0.85 g, 69%).
- Step 2 tert-butyl (1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-benzo[d]imidazol- 2-yl)cyclobutyl)carbamate (1 b)
- the mixture was subjected to microwave at 120 °C for 45 min and the reaction mass was extracted with ethyl acetate and washed with water.
- the ethyl acetate extracts were dried over Na 2 S0 4 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 2% MeOH/DCM) to yield a white foam (0.18 g, 54%).
- Step 3 tert-butyl 2-(8-(2-(1 -((tert-butoxycarbonyl)arriino)cyclobutyl)-1 H-benzo[d]irriidazol- 5-yl)-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (1 c)
- the mixture was subjected to microwave at 120 °C for 30 min. and the reaction mass was extracted with ethyl acetate and washed with water. The ethyl acetate extracts were dried over Na 2 S0 4 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 2-3% MeOH/DCM) to yield a yellowish white solid (130 mg, 84%).
- Step 4 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-((S)-1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-8-yl)- 1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 1 )
- Step 1 tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)carbamate (2a)
- the mixture was subjected to microwave at 1 15 °C for 45 min and the reaction mass was extracted with ethyl acetate and washed with water.
- the ethyl acetate extracts were dried over Na 2 S0 4 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 20-30% EtOAc/hexane) to yield an off white foam (0.20 g, 85%).
- Step 2 tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-5-yl)phenyl)-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (2b)
- Step 1 tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-5-yl)phenyl)-4,5-dihydro-3H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (3a)
- Step 1 (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (4a)
- Step 1 Synthesis of (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)- 1 H-benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (5a) NHBoc
- Step 1 tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6a)
- Step 2 tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6b)
- Step 3 Synthesis of (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)- 1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (6c)
- Step 4 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]i
- Step 1 (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (7a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2- d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide
- Step 1 (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (8a)
- Step 1 (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (9a).
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 9)
- Step 1 tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclopropyl)carbamate (10a)
- Step 2 tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopropyl)carbamate (10b)
- Step 3 (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopropyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (10c)
- Step 4 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol- 8-yl)phenyl)-1 -benzo[d]imidazol-2-yl)cyclopropyl)-3-methylbutanamide (Compound 10)
- Step 1 (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopropyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (1 1 a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2- d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopropyl)-3-methylbutanamide
- Step 1 tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12a)
- Step 2 tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H-benzo[d] imidazol-2-yl)cyclohexyl)carbamate (12b)
- Step 3 (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (12c)
- Step 1 (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (13a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2- d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide
- Step 1 (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl -1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (14a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 14):
- Step 1 (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (15a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-7- yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 15)
- Step 1 tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)(ethyl)carbamate (16a):
- Step 2 tert-butyl ethyl(1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)carbamate (16b):
- Step 3 (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)(ethyl)amino)cyclobutyl)-1 H-benzo [d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (16c):
- Step 1 tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)(methyl)carbamate (17a). Synthesized from 1 -((tert-butoxycarbonyl)(methyl)amino)cyclobutanecarboxylic acid [Synthesized according to procedure reported in WO 2010002655 A2] and 4- bromobenzene-1 ,2-diamine by following an analogous procedure described in Step 1 , Example 3.
- Step 2 tert-butyl methyl(1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)carbamate (17b)
- Step 3 (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)(methyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (17c)
- Example 20 Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N,3- dimethylbutanamide (Compound 18):
- Step 1 (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)(methyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (18a).
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)-N,3-dimethylbutanamide (Compound 18)
- Step 1 8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert-butoxycarbonyl) amino)cyclopentanecarboxylate (19a)
- Step 2 tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)cyclopentyl)carbamate (19b)
- Step 4 (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)-
- Step 2 tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)cyclohexyl)carbamate (20b)
- Step 4 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanamido)cyclohexyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol ⁇ yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 20)
- Step 1 Compoun
- Step 1 Compoun
- Step 1 tert-butyl (1 -(5-(5-bromothiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl) carbamate (23a)
- Step 2 (S)-tert-butyl 2-(7-(5-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H-benzo[d] imidazol-6-yl)thiophen- -yl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (23b)
- Step 3 (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)thiophen-2-yl)-1 H- benzo[d]imidazol- -yl)cyclobutyl)-3-methylbutanamide (Compound 23)
- Step 1 tert-butyl (1-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-benzo[d]imidazol- 2-yl)cyclohexyl)carbamate (24a).
- Step 3 (S)-tert-butyl 2-(7-(5-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)thiophen-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imid
- Step 1 (S)-tert-butyl 2-(7-((trimethylsilyl)ethynyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (2
- the reaction mixture was extracted with ethyl acetate and washed with water, and the organic layer was dried with Na 2 S0 4 and concentrated.
- the reddish oil was purified on CombiflashTM using 20% EtOAc-Hexane to obtain the pure compound (800 mg, 90%).
- Step 2 (S)-tert-butyl 2-(7-ethynyl-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine- 1 -carboxylate (25b).
- Step 3 (S)-tert-butyl 2-(7-((2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)ethynyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (25c)
- reaction mixture was extracted with ethyl acetate and washed with water, and the organic layer was dried over Na 2 S0 4 and concentrated.
- the reddish oil was purified on CombiflashTM with 20% EtOAc-Hexane to obtain pure compound (105 mg, 49%).
- Example 28 Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)ethyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 26):
- Step 4 (S)-tert-butyl 2-(7-(2-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)ethyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (26a) NHBoc
- Step 1 tert-butyl (1 -(5-(thiazol-2- -1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (27a)
- Step 3 Synthesis of (S)-tert-butyl 2-(8-(2-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)- 1 H-benzo[d]imidazol-5-yl)thiazol-5-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (27c)
- Step 1 (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-4,5-dihydro- 1 H-benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (28a)
- the mixture was subjected to microwave at 120 °C for 30 min. and the reaction mass was extracted with ethyl acetate and washed with water. The ethyl acetate extracts were dried over Na 2 S0 4 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 2-3% MeOH/DCM) to yield a yellowish white solid (60 mg, 28%).
- Step 1 (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (29a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepin -d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 29)
- Step 1 6-bromo-1 -oxo-1 ,2,3,4-tetrahydronaphthalen-2-yl 1 -((tert-butoxycarbonyl)amino) cyclobutanecarboxylate (30a) Synthesized from 2,6-dibromo-3,4-dihydronaphthalen-1 (2H)-one and 1 -((tert- butoxycarbonyl)amino)cyclobutanecarboxylic acid by following an analogous procedure described in Step 1 , Example 21 .
- Step 2 tert-butyl (1 -(7-bromo-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl) carbamate (30b)
- Step 3 (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (30c)
- Example 33 Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide
- Step 1 (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl -1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (31 a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-1 H- naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 31 )
- Step 1 6-bromo-1 -oxo-1 ,2,3,4-tetrahydronaphthalen-2-yl 1 -((tert-butoxycarbonyl)amino) cyclohexanecarboxylate (32a)
- Step 2 tert-butyl (1-(7-bromo-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl) carbamate (32b)
- Step 3 (S)-tert-butyl 2-(6-(4-(2-(1-((tert-butoxycarbonyl)amino)cyclohexyl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (32c)
- Step 1 (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (33a)
- Step 2 (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-1 H- naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 33)
- Example 36 Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide (Compound 34):
- Step 1 8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert-butoxycarbonyl) amino)cyclobutanecarboxylate
- Step 2 tert-butyl (1 -(8-bromo-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl) cyclobutyl)carbamate (34b)
- Step 3 (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (34c)
- Anti-viral activity of the compounds of the invention was monitored using an HCV replicon assay.
- the Huh7.5/ Con1/SG-Neo(l)hRluc2aUb cell line persistently expressing a bicistronic genotype 1 b replicon in Huh 7.5 cells was obtained from Apath LLC. This cell line was used to test inhibition of replicon levels by test compound using Renilla luciferase enzyme activity readout as a measure of viral replication efficiency.
- Table 1 shows EC 5 o values, for inhibition of genotype 1 b replicon, of the compounds of the invention.
- Group A compounds exhibited EC 50 value between 1 pM to 499 pM
- Group B exhibited EC 5 o value between 500 pM to 999 pM
- Group C exhibited EC 50 value of more than 1 nM.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compounds of the general formula (I), their tautomeric forms, their stereoisomers, their analogs, their prodrugs, their isotopes, their N-oxides, their metabolites, their pharmaceutically acceptable salts, polymorphs, solvates, optical isomers, clathrates, co-crystals, combinations with suitable medicament, pharmaceutical compositions containing them, methods of making of the above compounds, and their use as antiviral candidate, more specifically as anti-HCV are disclosed.
Description
ANTIVIRAL COMPOUNDS
FIELD OF THE INVENTION:
The present invention is related to novel compounds of the general formula I,

their tautomeric forms, their stereoisomers, their analogs, their prodrugs, their isotopes, their N-oxides, their metabolites, their pharmaceutically acceptable salts, polymorphs, solvates, optical isomers, clathrates, co-crystals, combinations with suitable medicament, pharmaceutical compositions containing them, methods of making of the above compounds , and their use as antiviral candidate, more specifically as anti-HCV. BACKGROUND OF THE INVENTION
Persistent hepatitis C virus (HCV) infection is a major health problem globally affecting -3% of the world population and is an important contributor to chronic liver disease culminating with liver cirrhosis, hepatocellular carcinoma and liver failure [Szabo E,Lotz G, et al., Pathol. Oncol. Res. 2003, 9, 215-221 ; Hoofnagle JH., Hepatology 1997, 26 15S-20]. An estimated 170 million chronic carriers worldwide are at risk of developing liver disease. In the United States alone ~3 million are chronically infected with HCV and the number of HCV related deaths is increasing significantly over the years [Barnes E., WHO factsheet 2010. Available at: http://www.who.int/vaccine_research/diseases/viral_cancers/en/index2.html]. Clinically, chronic infection is often asymptomatic with latent periods lasting for decades before manifestation by which time extensive liver damage has occurred. HCV is spread primarily by unscreened blood transfusions and use of contaminated needles and syringes; the highest risk groups are intravenous drug users and people who received blood transfusions (mainly haemophiliacs) before 1990 when screening for HCV was introduced. Factors that have been reported to influence the rate of HCV disease progression include age (increasing age is associated with more rapid progression), gender (males have more rapid disease progression than females), alcohol consumption (associated with an
increased rate of disease progression), HIV co-infection (associated with a markedly increased rate of disease progression), and fatty liver.
Despite significant efforts, no vaccine exists for HCV and until a year ago the standard therapy for HCV was a combination of pegylated interferon (PEG-IFN) a and weight based ribavarin (RBV), which was inadequate for majority of the patients and therapy associated side effects such as pancytopenia, flu-like symptoms or depression were commonly observed leading to early treatment discontinuation [Fried MW, et al., N Engl J Med. 2002, 347, 975-982]. The approval of two direct acting agents (DAA) i.e. 1 st generation protease inhibitors, Incivek and Victrelis in May 201 1 ushered in the era of specifically targeted HCV therapy[Jesudian AB, Gambarin-Gelwan M and Jacobson IM., Gastroenterology Hepatol. 2012, 8, 91 -101 .
The combination of above mentioned DAAs, Peg-IFN and RBV (triple therapy) substantially increases the rate of sustained virologic response in treatment nal've and experienced patients. However, a number of issue restrict the usage of these drugs - i) complex treatment algorithms issued by the regulatory bodies; ii) they are restricted to genotype 1 ; iii) low barrier to resistance mutations and iv) increased cost of therapy leading to limited access to care. Hence, there exists a need for alternative therapeutic strategies that ensure broader genotype coverage, better efficacy, better tolerance and limited selection of resistant HCV variants. The sequence diversity of HCV is complex with the virus organized into 6 distinct genotypes and over 100 subtypes. Additionally, HCV exists as many closely related viral sequences, termed as quasi-species, in the infected individual, making specific pharmaceutical targeting of HCV proteins challenging due to the rapid evolution of escape mutants. It is increasingly evident that a broad collection of specific, pan genotypic anti- viral drugs targeting multiple essential viral functions, in addition to the current viral therapies will be required for effective global control of HCV.
Disclosures describing the synthesis of HCV inhibitors are US20090202478, US20090202483, WO2009020828, WO2009020825, WO2009102318, WO2009102325, WO2009102694, WO2008144380, WO2008021927, WO2008021928, WO2008021936, WO2006133326, WO2004014852, WO2008070447, WO2009034390, WO2006079833, WO2007031791 , WO2007070556, WO2007070600, WO2008064218, WO2008154601 ,
WO2007082554, WO2008048589, EP2121697, US8008264, US8008263, US201 10217265, US201 10217261 , US8012982, US8012942, US8012941 , US201 10223134, WO201 1 106992, WO201 1 106929, US201 10237636, US201 10237579, US201 10236348, US201 10250176, US201 10250172, US201 1269956, US201 1274648, EP2385048, US201 10281910, US201 10286961 , US201 10294819, US201 10293563, US201 1300104, WO201 1 156543, WO201 1 153396, WO201 1 151652, WO201 1 151651 , US2012004196, US8093243, US8101643, US20120028978, WO2012018534, WO2012018325, WO2012021704, WO2012021591 , US20120040977, US20120040962, WO2012024363, ΕΡ2086995, ΕΡ20491 16, US8133884, US20120076755, ΕΡ2250163, US8143414, US8143301 , US8143288, US20120083483, US8147818, WO2012039717, WO2012041227, WO2012041014, ΕΡ2146984, US8188132, and US8198449.
SUMMARY OF THE INVENTION
According to one aspect of the present invention there is provided a novel compound of the general formula (I), its tautomeric forms, its stereoisomers, its analogs, its prodrugs, its isotopes, its N-oxides, its metabolites, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates, or its co-crystals,

HCV is a member of the Flaviviridae family of enveloped, positive stranded RNA viruses belonging to the genus Hepacivirus. The genome is a single ~9.6kb strand of RNA and consists of one open reading frame that encodes for a polyprotein of -3000 amino acids flanked by untranslated regions at both 5' and 3' ends. This precursor polyprotein is then processed by viral and cellular proteases to yield 10 separate mature viral proteins critical for replication and assembly of progeny viral particles. The organization of the structural and non-structural proteins in the HCV polyprotein is as follows: C-E1 -E2-P7-NS2-NS3- NS4a-NS4b-NS5a-NS5b. The three structural proteins C, E1 and E2 are involved in packaging of the virus and the infectivity cycle. The function of the p7 protein is unknown. Of the non-structural proteins, NS2 is a zinc dependent metalloproteinase that functions in conjunction with a part of NS3 protein. NS3 protein has two catalytic activities associated with it: a serine protease at the N-terminal end which requires NS4A as a cofactor, and an ATPase dependent helicase activity at the C-terminal end. NS5A is a membrane anchored phosphoprotein that is present in basally phosphorylated (56kDa) and hyperphosphorylated (58kDa) forms. Its precise role has not been determined but it has been shown to play a role in RNA binding, multiple host protein interactions, and interferon resistance. Additionally recent evidence suggests that NS5A plays an important role in replication and infectivity of HCV. The NS5B protein encodes an RNA dependent RNA polymerase activity, key to the generation of progeny viruses. While the pathology of HCV infection mainly affects the liver, the virus is found in other cell types in the body including peripheral blood lymphocytes [Thomson BJ et al., Clin Microbial Infect. 2005, V\_, 86-94; Moriishi K et al., Antivir. Chem. Chemother. 2003, 14, 285-297]. Characterization of the replicase machinery required for HCV RNA synthesis has defined the protease/helicase NS3 protein, the NS4A cofactor, the NS4B integral membrane protein, the NS5A protein and the RNA dependent RNA polymerase NS5B as being its essential components.
Hence, one of the aspects of the present invention is provision of novel compounds of the general formula I,


Ring 'A' is selected from 3 to 6 membered carbocycle and 3 to 6 membered heterocycle, the ring 'A' may further be annulated with substituted- or unsubstituted- carbocycle, substituted- or unsubstituted- heterocycle, substituted- or unsubstituted- aromatic carbocycle or substituted- or unsubstituted- aromatic heterocycle; ring D is selected from substituted- or unsubstituted- 5 to 10 membered carbocycles, substituted- or unsubstituted- 5 to 10 membered heterocycles containing 1 to 3 heteroatoms/groups selected from N(R10), S(0)p, O or C(=0), substituted- or unsubstituted- aromatic carbocycle, and 5 to 6 membered substituted- or unsubstituted- aromatic heterocycle containing heteroatoms selected from N, S or O;
Ring Έ' is 5 to 10 membered heterocycle, the ring Έ' may be monocyclic, fused bicyclic, bridged bicyclic or spiro bicyclic;
R1 is selected independently at each occurrence from the group consisting of halogen, substituted- or unsubstituted- Ci-6 alkyl, R10O-, R10aOC(=O)-, and (R10)(R11)NC(=O)-; or two R1s taken together may form an oxo (=0) group;
R2 is selected independently at each occurrence from the group consisting of substituted- or unsubstituted- Ci-6 alkyl, R10aC(=O)-, R10aSO2-, R10aOC(=O)-, R10(R11)NC(=O)-, R10aOC(=O)N(R11)C(Ra)(Rb)C(=O)-, R10aOC(=O)N(R11)C(Ra)(Rb)C(Rc)(Rd)C(=O)-, R10(R11)NC(=O)N(R12)C(Ra)(Rb)C(=O)-, R10(R11)NC(=O)N(R12)C(Ra)(Rb)C(Rc)(Rd)C(=O)-,
R10aSO2N(R11)C(Ra)(Rb)C(=O)-, R10aSO2N(R11)C(Ra)(Rb)C(Rc)(Rd)C(=O)-, and R10aOC(=O)N(R11)C(Ra)(Rb)SO2-;
R3 is selected from O and N(R13);
R4 is selected independently at each occurrence form CRa(Rb), O, N(R13), C(=0), S(0)p, substituted- or unsubstituted carbocycle, substituted- or unsubstituted- heterocycle, substituted- or unsubstituted- arylene and substituted- or unsubstituted- heteroarylene, wherein, substitutions on carbocycle, heterocycle, arylene, and heteroarylene are selected from the group consisting of halogen, substituted- or unsubstituted- Ci-6 alkyl, and alkyl-O-; R5 is selected from hydrogen and substituted- or unsubstituted- alkyl;
R6 and R7 are independently selected from the group consisting of hydrogen, halogen, substituted- or unsubstituted- Ci-6 alkyl, and R10O-;
R8 is selected from hydrogen and Ci-6 alkyl;
R9 is selected from the group consisting of hydrogen, Ci-6 alkyl, R10aC(=O)-, R10aSO2-, R10aOC(=O)-, (R10)(R11)NC(=O)-, R10aOC(=O)N(R11)CRa(Rb)C(=O)-,
R10aOC(=O)N(R11)CRa(Rb)C(Rc)(Rd)C(=O)-, R10(R11)NC(=O)N(R12)CRa(Rb)C(=O)-, R10(R11)NC(=O)N(R12)CRa(Rb)C(Rc)(Rd)C(=O)-, R10aSO2N(R11)CRa(Rb)C(=O)-, R10aSO2N(R11)CRa(Rb)C(Rc)(Rd)C(=O)-, and R10aOC(=O)N(R11)CRa(Rb)SO2-;
R10, R11 , R11a and R12 are independently selected from hydrogen and substituted- or unsubstituted- Ci-6 alkyl;
R10a is substituted- or unsubstituted- Ci-6 alkyl;
R13 is selected from hydrogen and substituted- or unsubstituted- alkyl group;
Ra, Rb, Rc and Rd, are independently selected from hydrogen, halogen, substituted- or unsubstituted- Ci-6 alkyl, substituted- or unsubstituted- aryl, substituted- or unsubstituted- heteroaryl, substituted- or unsubstituted- cycloalkyl, and substituted- or unsubstituted- heterocyclyl, or Ra, Rb, Rc and Rd together with the carbon atom(s) to which they are
attached forming substituted- or unsubstituted- carbocycle, substituted- or unsubstituted- heterocycle; m is an integer ranging between 0 to 2, selected independently at each occurrence; n is an integer ranging between 0 and 2; p is an integer ranging between 0 and 2; when n = 2 and R4 is selected as CRa(Rb) for both the occurrences, two Ras together can form a bond to form a alkenylene linkage or two Ras and two Rbs together can form bonds to form alkynylene linkage;
'alkyl' may be substituted with 1 to 4 substituents selected from the group consisting of oxo, halogen, cyano, aryl, hereroaryl, cycloalkyl, heterocyclyl, (alkyl)C(=0)-, (alkyl)S02-, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, (R14)(H)NC(=0)-, (R14)(alkyl)NC(=0)-, (alkyl)C(=0)N(H)-, R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-
'cycloalkyl' and 'carbocycle' may be substituted with 1 to 2 substituents selected from the group consisting of oxo, halogen, Ci-6 alkyl, haloalkyl, (alkyl)C(=0)-, (alkyl)S02-, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, R14(H)NC(=0)-, R14(alkyl)NC(=0)-, (alkyl)C(=0)N(H)-, R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-;
'aryl' or 'aromatic carbocycle', may be substituted with 1 to 2 substituents selected from the group consisting of halogen, nitro, cyano, hydroxy, Ci to C6 alkyl, perhaloalkyl, alkyl-O- , perhaloalkyl-O-, alkyl-N(alkyl)-, alkyl-N(H)-, H2N-, alkyl-S02-, alkyl-C(=0)N(alkyl)-, alkyl- C(=0)N(H)-, alkyl-N(alkyl)C(=0)-, alkyl-N(H)C(=0)-, H2NC(=0)-, alkyl-N(alkyl)S02-, alkyl- N(H)S02-, H2NS02-;
'heteroaryl' or 'aromatic heterocycle' may be substituted with 1 to 2 substituents selected from the group consisting of halogen, nitro, cyano, hydroxy, Ci to C6 alkyl, perhaloalkyl, alkyl-O-, perhaloalkyl-O-, alkyl-N(alkyl)-, alkyl-N(H)-, H2N-, alkyl-S02-, alkyl-C(=0)N(alkyl)-, alkyl-C(=0)N(H)-, alkyl-N(alkyl)C(=0)-, alkyl-N(H)C(=0)-, H2NC(=0)-, alkyl-N(alkyl)S02-, and alkyl-N(H)S02-, H2NS02-; ring carbons of 'heterocyclyl' and 'heterocycle' may be substituted with 1 to 2 substituents selected from the group consisting of oxo, halogen, alkyl, R14aO-, (alkyl)OC(=0)-,
(alkyl)C(=0)0-, R14(H)NC(=0)-, R14(alkyl)NC(0)-, (alkyl)C(=0)N(H)-, R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-; the substituents on ring nitrogen(s) of 'heterocyclyl' and 'heterocycle' are selected from the group consisting of alkyl, (alkyl)C(=0)-, (alkyl)S02-, (alkyl)OC(=0)-, R14(H)NC(=0)-, R14(alkyl)NC(=0)-; R14 is selected from hydrogen and alkyl ;
R14a is selected from hydrogen, alkyl, perhaloalkyl.
Whenever a range of the number of atoms in a structure is indicated (e.g., a C1 -12, Ci-8, Ci-
6, or C-1 -4 alkyl, alkylamino, etc.), it is specifically contemplated that any sub-range or individual number of carbon atoms falling within the indicated range also can be used. Thus, for instance, the recitation of a range of 1 -8 carbon atoms (e.g., Ci-C8), 1 -6 carbon atoms (e.g., Ci-C6), 1 -4 carbon atoms (e.g., CrC4), 1 -3 carbon atoms (e.g., Ci-C3), or 2-8 carbon atoms (e.g., C2-C8) as used with respect to any chemical group (e.g., alkyl, alkylamino, etc.) referenced herein encompasses and specifically describes 1 , 2, 3, 4, 5, 6,
7, 8, 9, 10, 1 1 , and/or 12 carbon atoms, as appropriate, as well as any sub-range thereof (e.g., 1 -2 carbon atoms, 1 -3 carbon atoms, 1 -4 carbon atoms, 1 -5 carbon atoms, 1 -6 carbon atoms, 1 -7 carbon atoms, 1 -8 carbon atoms, 1 -9 carbon atoms, 1 -10 carbon atoms, 1 -1 1 carbon atoms, 1 -12 carbon atoms, 2-3 carbon atoms, 2-4 carbon atoms, 2-5 carbon atoms, 2-6 carbon atoms, 2-7 carbon atoms, 2-8 carbon atoms, 2-9 carbon atoms, 2-10 carbon atoms, 2-1 1 carbon atoms, 2-12 carbon atoms, 3-4 carbon atoms, 3-5 carbon atoms, 3-6 carbon atoms, 3-7 carbon atoms, 3-8 carbon atoms, 3-9 carbon atoms, 3-10 carbon atoms, 3-1 1 carbon atoms, 3-12 carbon atoms, 4-5 carbon atoms, 4-6 carbon atoms, 4-7 carbon atoms, 4-8 carbon atoms, 4-9 carbon atoms, 4-10 carbon atoms, 4-1 1 carbon atoms, and/or 4-12 carbon atoms, etc., as appropriate). One of the embodiments of the present invention is compound of formula (I) as described above, wherein, RA is selected independently at each occurrence from -


In any of the embodiments of the invention described above, R3 is particularly selected as NH.
In any of the embodiments of the invention described above, R4 is particularly selected from phenylene and five membered heteroarylene containing 1 or 2 heteroatoms selected from N and S, when n is particularly selected as 1 .
In any of the embodiments of the invention described above, R4 is particularly selected as CRaRb, when n is particularly selected as 2 to form an ethylene or ethynylene linkage.
In any of the embodiments of the invention described above, n is particularly selected from 0, 1 and 2;
In any of the embodiments of the invention described above, R5, R6 and R7 are each particularly selected as hydrogen.
In any of the embodiments of the invention described above, Ring D is particularly selected from the group consisting of aromatic carbocycle, six membered carbocycle, seven membered carbocycle, and seven membered heterocycle containing one heteroatom particularly selected as oxygen.
In any of the embodiments of the invention described above, RA is selected independently at each occurrence from -


General terms used in formula can be defined as follows; however, the meaning stated should not be interpreted as limiting the scope of the term per se.
The term "alkyl", as used herein, means a straight chain or branched hydrocarbon containing from 1 to 20 carbon atoms. Preferably the alkyl chain may contain 1 to 10 carbon atoms. More preferably alkyl chain may contain up to 6 carbon atoms. Representative examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, and n- hexyl.
'Alkyl' as defined hereinabove may be substituted with one or more substituents selected independently from the group comprising of oxo, halogen, cyano, aryl, hereroaryl, cycloalkyl, heterocyclyl, (alkyl)C(=0)-, (alkyl)S02-, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, (R14)(H)NC(=0)-, (R14)(alkyl)NC(=0)-, (alkyl)C(=0)N(H)-, R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-; wherein, R14 is selected from hydrogen and alkyl, R14a is selected from hydrogen, alkyl, perhaloalkyl;
The term "haloalkyl" used herein means an alkyl group as defined hereinabove wherein at least one of the hydrogen atoms of the said alkyl group is substituted with halogen. The haloalkyl group is exemplified by monofluoromethyl, 1 ,2-dichloroethyl and the like. The term "perhaloalkyl" means an alkyl group as defined hereinabove wherein all the
hydrogen atoms of the said alkyl group are substituted with halogen. The perhaloalkyl group is exemplified by trifluoromethyl, pentafluoroethyl and the like.
The term "cycloalkyi" as used herein, means a monocyclic, bicyclic, or tricyclic non- aromatic ring system containing from 3 to 14 carbon atoms, preferably monocyclic cycloalkyi ring containing 3 to 6 carbon atoms. Examples of monocyclic ring systems include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl. Bicyclic ring systems are also exemplified by a bridged monocyclic ring system in which two non-adjacent carbon atoms of the monocyclic ring are linked by an alkylene bridge. Representative examples of bicyclic ring systems include, but are not limited to, bicyclo[3.1 .1 ]heptane, bicyclo[2.2.1 ]heptane, bicyclo[2.2.2]octane, bicyclo[3.2.2]nonane, bicyclo[3.3.1 ]nonane, and bicyclo[4.2.1 ]nonane, bicyclo[3.3.2]decane, bicyclo[3.1 .0]hexane, bicyclo[410]heptane, bicyclo[3.2.0]heptanes, octahydro-1 H-indene. Tricyclic ring systems are also exemplified by a bicyclic ring system in which two non- adjacent carbon atoms of the bicyclic ring are linked by a bond or an alkylene bridge. Representative examples of tricyclic-ring systems include, but are not limited to, tricyclo[3.3.1 .03 7]nonane and tricyclo[3.3.1 .13 7]decane (adamantane). The term cycloalkyi also include spiro systems wherein one of the ring is annulated on a single carbon atom such ring systems are exemplified by spiro[2.5]octane, spiro[4.5]decane, spiro[bicyclo[4.1 .0]heptane-2,1 '-cyclopentane], hexahydro-2'H-spiro[cyclopropane-1 , 1 '- pentalene].
The term "cycloalkenyl" as used herein, means a cycloalkyi group as defined above containing at least one double bond.
The term "carbocycle" as used herein, means a cyclic system made up of carbon atoms, which includes cycloalkane, cycloalkene and aromatic carbocycle.
Cycloalkyi, cycloalkenyl and carbocycle as defined hereinabove may be substituted with one or more substituents selected independently from the group comprising of oxo, halogen, Ci-6 alkyl, haloalkyl, (alkyl)C(=0)-, (alkyl)S02-, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, R14(H)NC(=0)-, R14(alkyl)NC(=0)-, (alkyl)C(=0)N(H)-, R14(H)N-,
R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-; wherein, R14 is selected from hydrogen and alkyl, R14a is selected from hydrogen, alkyl, perhaloalkyl.
The term "aryl" refers to a monovalent monocyclic, bicyclic or tricyclic aromatic hydrocarbon ring system. Examples of aryl groups include phenyl, naphthyl, anthracenyl, fluorenyl, indenyl, azulenyl, and the like. Aryl group also include partially saturated bicyclic and tricyclic aromatic hydrocarbons such as tetrahydro-naphthalene. The said aryl group also includes aryl rings fused with heteroaryl or heterocyclic rings such as 2,3-dihydro- benzo[1 ,4]dioxin-6-yl, 2,3-dihydro-benzo[1 ,4]dioxin-5-yl, 2,3-dihydro-benzofuran-5-yl, 2,3- dihydro-benzofuran-4-yl, 2,3-dihydro-benzofuran-6-yl, 2,3-dihydro-benzofuran-6-yl, 2,3- dihydro-1 H-indol-5-yl, 2,3-dihydro-1 H-indol-4-yl, 2,3-dihydro-1 H-indol-6-yl, 2,3-dihydro-1 H- indol-7-yl, benzo[1 ,3]dioxol-4-yl, benzo[1 ,3]dioxol-5-yl, 1 ,2,3,4-tetrahydroquinolinyl, 1 ,2,3,4-tetrahydroisoquinolinyl, 2,3-dihydrobenzothien-4-yl, 2-oxoindolin-5-yl.
Aryl as defined hereinabove may be substituted with one or more substituents selected independently from the group consisting of halogen, nitro, cyano, hydroxy, Ci to C6 alkyl, perhaloalkyl, alkyl-O-, perhaloalkyl-O-, alkyl-N(alkyl)-, alkyl-N(H)-, H2N-, alkyl-S02-, alkyl- C(=0)N(alkyl)-, alkyl-C(=0)N(H)-, alkyl-N(alkyl)C(=0)-, alkyl-N(H)C(=0)-, H2NC(=0)-, alkyl-N(alkyl)S02-, alkyl-N(H)S02-, H2NS02-.
The term "heteroaryl" refers to a 5- 14 membered monocyclic, bicyclic, or tricyclic ring system having 1 -4 ring heteroatoms selected from O, N, or S, and the remainder ring atoms being carbon (with appropriate hydrogen atoms unless otherwise indicated), wherein at least one ring in the ring system is aromatic. Heteroaryl groups may be optionally substituted with one or more substituents. In one embodiment, 0, 1 , 2, 3, or 4 atoms of each ring of a heteroaryl group may be substituted by a substituent. Examples of heteroaryl groups include pyridyi, 1 -oxo-pyridyl, furany!, thienyl, pyrroiyL oxazolyl, oxadiazolyl, imidazolyl, thiazolyl, isoxazolyl, quinolinyl, pyrazolyl, isothiazolyl, pyridazinyL pyrimidinyl, pyrazinyl, triazinyl. triazolyl. thiadiazolyl, isoquinolinyl, benzoxazolyl, benzofuranyl, indolizinyl, imidazopyridyl, tetrazolyl, benzimidazolyi, benzothiazolyl, benzothiadiazolyl, benzoxadiazolyl, indolyl, azaindolyL imidazopyridyl, quinazolinyl, purinyl, pyrrolo[2,3]pyrimidinyi, pyrazolo[3,4]pyrimidinyl, and benzo(b)thienyi, 2,3-
thiadiazolyL 1 H-pyrazoio[5,1 -c]-1 ,2,4-triazoiyi, pyrro!o[3,4-d]-1 ,2,3-triazolyi, cyclopentatriazolyl, 3H-pyrrolo[3,4-c] isoxazoly! and the like.
Heteroaryl as defined hereinabove may be substituted with one or more substituents selected independently from the group consisting of halogen, nitro, cyano, hydroxy, Ci to C6 alkyl, perhaloalkyl, alkyl-O-, perhaloalkyl-O-, alkyl-N(alkyl)-, alkyl-N(H)-, H2N-, alkyl- S02-, alkyl-C(=0)N(alkyl)-, alkyl-C(=0)N(H)-, alkyl-N(alkyl)C(=0)-, alkyl-N(H)C(=0)-, H2NC(=0)-, alkyl-N(alkyl)S02-, and alkyl-N(H)S02-, H2NS02-.
The term "heterocycle" or "heterocyclic" as used herein, means a 'cycloalkyi' group wherein one or more of the carbon atoms replaced by -0-, -S-, -S(02)-, -S(O)-, -N(Rm)-, - Si(Rm)Rn-, wherein, Rm and Rn are independently selected from hydrogen, alkyl, aryl, heteroaryl, cycloalkyi, and heterocyclyl. The heterocycle may be connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the heterocycle. Representative examples of monocyclic heterocycle include, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1 ,3-dioxanyl, 1 ,3-dioxolanyl, 1 ,3- dithiolanyl, 1 ,3-dithianyl, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl. oxadiazolidinyl, oxazolinyl, oxazolidinyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl. pyrrolinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydrothienyl, thiadiazolinyl, thiadiazolidinyl, thiazolinyl, thiazolidinyl, thiomorpholinyl, 1 .1 -dioxidothiomorpholinyl (thiomorpholine sulfone). thiopyranyl, and trithianyl. Representative examples of bicyclic heterocycle include, but are not limited to 1 ,3-benzodioxolyl, 1 ,3-benzodithiolyl, 2,3-dihydro-1 ,4-benzodioxinyl, 2,3- dihydro-1 -benzofuranyl, 2,3-dihydro-1 -benzothienyl, 2,3-dihydro-1 H-indolyl and 1 ,2,3,4- tetrahydroquinolinyl. The term heterocycle also include bridged heterocyclic systems such as azabicyclo[3.2.1 ]octane, azabicyclo[3.3.1 ]nonane and the like.
Heterocyclyl group may be substituted on ring carbons with one or more substituents selected independently from the group consisting of oxo, halogen, alkyl, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, R14(H)NC(=0)-, R14(alkyl)NC(0)-, (alkyl)C(=0)N(H)-, R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-; wherein, R14 is selected from hydrogen and alkyl, R14a is selected from hydrogen, alkyl, perhaloalkyl.
Heterocyclyl group may further be substituted on ring nitrogen(s) with substituents selected from the group consisting of alkyl, (alkyl)C(=0)-, (alkyl)S02-, (alkyl)OC(=0)-, R14(H)NC(=0)-, R14(alkyl)NC(=0)-; wherein, R14 is selected from hydrogen and alkyl, R14a is selected from hydrogen, alkyl, perhaloalkyl.
The term Όχο' means a divalent oxygen (=0) attached to the parent group. For example oxo attached to carbon forms a carbonyl, oxo substituted on cyclohexane forms a cyclohexanone, and the like. The term 'annulated' means the ring system under consideration is either annulated with another ring at a carbon atom of the cyclic system or across a bond of the cyclic system as in the case of fused or spiro ring systems.
The term 'bridged' means the ring system under consideration contain an alkylene bridge having 1 to 4 methylene units joining two non-adjacent ring atoms.
A compound its stereoisomers, racemates, pharmaceutically acceptable salt thereof as described hereinabove wherein the compound of general formula I is selected from:
1 . (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-((S)-1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol- 8-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide. (Compound 1 )
2. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)- 3-methylbutanamide. (Compound 2)
3. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide. (Compound 3) 4. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-
tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclobutyl)-3-methylbutanamide. (Compound 4) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide. (Compound 5) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopentyl)-3-methylbutanamide. (Compound 6) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopentyl)-3-methylbutanamide. (Compound 7) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide. (Compound 8) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide. (Compound 9) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopropyl)-3-methylbutanamide. (Compound 10) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopropyl)-3-methylbutanamide. (Compound 1 1 )
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide. (Compound 12) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide. (Compound 13) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide. (Compound 14) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide. (Compound 15) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N-ethyl-3- methylbutanamide. (Compound 16) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N,3- dimethylbutanamide. (Compound 17) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N,3- dimethylbutanamide. (Compound 18) (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)-3- methylbutanamido)cyclopentyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-
benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide.
(Compound 19) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)-3- methylbutanamido)cyclohexyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-8- yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide. (Compound 20) (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)-3- methylbutanamido)cyclohexyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide. (Compound 21 ) (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)-3- methylbutanamido)cyclohexyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide.
(Compound 22) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)thiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide. (Compound 23) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)thiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide. (Compound 24) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-((2-((S)-1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-7- yl)ethynyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide. (Compound 25) (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)ethyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide. (Compound 26)
(S)-2-(methoxycarbonyl)amino-N-(1 -(5-(5-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)thiazol-2-yl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide. (Compound 27) (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6 yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide. (Compound 28) (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6 yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide. (Compound 29) (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6 yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide. (Compound 30) (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6 yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide.
(Compound 31 ) (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6 yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide. (Compound 32) (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6 yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide.
(Compound 33) (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2-
((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6
yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide. (Compound 34)
According to a feature of the present invention, the compounds of general formula I where all the symbols are as defined earlier, can be prepared by methods given in Schemes given below or in the examples; the disclosure should not be construed to limit the scope of the invention arriving at compound of formula I as disclosed hereinabove.
Ring A as provided in compound of formula T is alternatively represented as follows -
 where, m = 1 -2; n = 0-2; X = N(R10), O, CH2, or C(=0)
where, m = 1 -2; n = 0-2; X = N(R10), O, CH2, or C(=0)
Scheme 1 :

The benzimidazole derivatives 4 could be prepared by coupling different ring sized cyclic (carbocyclic or heterocyclic) amino acids 2 with 1 ,2-diamino-4-bromobenzene 1 . The coupling of 1 with Boc-protected amino acid 2 is carried out either by using conventional coupling reagents such as EDCI or HATU could provide a mixture of amide derivatives 3, Scheme 1 . Alternatively, 3 could be prepared by the conversion of the protected amino acids to acid chloride by isobutyl chloroformate in the presence of a base and subsequent addition to 1 ,2-diamino-4-bromobenzene in the presence of an organic base such as Et3N or N-methylmorpholine. The mixture of regio isomers can then be cyclised to the benzimidazole derivative 4 in presence of acetic acid or P205 or by simple reflux with
ethanol for extended periods. The bromobenzimidazole derivative 4 or its boronate ester 5, prepared by Pd catalysed methods, used in further coupling reactions with the tricyclic moiety.
The tricyclic units of different ring size 6 can be synthesized by a number of possible methods from the easily accessible tetralone moiety 7 as provided in WO2009102633.
The alpha-bromo ketone 8, synthesized by addition of molecular bromine to the tetralone moiety 7, on O-Alkylation with an amino acid such as N-Boc proline could provide intermediate 9 which can be cyclised to the imidazole containing tricycle 6 using NH4OAc.
Alternatively, the oximino-ketone 10, prepared by the addition of alkyl nitrite to the tetralone moiety 7, can be cyclised with an aldehyde such as N-Boc-Prolinal in the presence of ammonium acetate to the hydroxylated imidazole derivative 11 using the process provided in Bioorg. Med. Chem. Lett., 2002, 1009-101 1 . Treatment of 11 with triethyl phosphite can provide the imidazole containing tricycle 6.
Another approach could be from the alpha-amino ketone 13, which can be synthesized from the oximino-ketone 10 or the tetralone moiety 7, Scheme 2. Amide coupling with an N-Boc protected amino acid such as Proline could provide the amide 14 which can be cyclised to the imidazole containing tricycle 6 in the presence of ammonium acetate.

The tricyclic moiety 6 or it's boronate ester 17 used in Suzuki reaction to couple with the benzimidazole unit 5 or 4, respectively to provide the coupled product 18, Scheme 3, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 3:

Compounds of formula I with a linker (R4) group such as phenyl or heteroaryl between the benzimidazole and tricyclic unit can be prepared by the Suzuki reaction (Chem. Rev., 1995, 95, 2457-83) of bromo benzimidazole derivative 4 or tricyclic bromide 6 with excess of 1 ,4-bis(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzene or the related heterocyclic bisboronate to provide the corresponding boronates 19 or 20, respectively, Scheme 4. Suzuki reaction between the benzimidazole boronate 19 and the tricyclic bromides 6 or coupling of 20 and 4 would give rise to compounds of formula 21 , which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 4:


X2,Y2. Z2 are independently selected from C and N
Compounds of formula I with R4 as five membered heteroaryl can be made by reacting the bromoheteroaryl compounds with the benzimidazole or tricyclic boronates 5 or 17 via the Suzuki reaction to provide the coupled products 22 or 23, respectively. Bromination with NBS would provide compounds 24 and 25. These bromides 24 or 25 can be coupled with the boronate 17 or 5, respectively to give rise to compounds such as 26 or 27 with a 5 membered aryl unit linking the tricycle and benzimidazole moieties, Scheme 5, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 5:

An alkyne linker can be introduced by the Sonogashira reaction (J. Org. Chem., 2006, 71 , 1 , 379-381 ) of TMS-acetylene with either of the bromides 4 or 6 and deprotecting the TMS group with K2C03 and a subsequent Sonogashira reaction with the alternate bromides, Scheme 6, (i.e 1 st Sonogashira with a benzimidazole bromide and 2nd Sonogashira with a tricyclic bromide and vice versa) would provide compounds such as 30, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 6:

The alkyne linker can be reduced to ethylene linker by catalytic hydrogenation to give compounds of formula 31 , Scheme 7, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 7:

Compounds with heterocycle such as piperazine as linker group (R4) can be prepared by a sequential Buchwald reaction (J. Am. Chem. Soc, 2003, 125, 22, 6653-55; Acc. Chem. Res., 1998, 31 , 852-860) of 1 -Cbz-Piperazine with bromide 4 or 6 followed by catalytic hydrogenation to remove the Cbz group and second Buchwald reaction with the alternate bromides 6 or 4, respectively, would give the intermediate 34, Scheme 8, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 8:

Compounds with linkers such as a -0-CH2- can be introduced by converting the bromo tricyclic unit 6 to a vinyl alkene by a Stille reaction (Canadian Journal of Chemistry, 2000, 78, 7, 957-962), and the alkene functionality can be transformed to an aldehyde by ozonolysis or by oxidative cleavage with sodium periodate in the presence of ruthenium or osmium catalysts, Scheme 9. The aldehyde can be reduced to an alcohol and the alcohol can be transformed to a suitable leaving group such as tosylate, mesylate, bromide, etc to give intermediate 35, which can be reacted with phenolic benzimidazoles 36 to give compound 37, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 9:

Compounds with linker such as -O- can be prepared by reaction of the phenolic benzimidazoles such as 36 with the tricyclic system 6 with appropriate halo substitution such as Br, CI, or F in the presence of a base such as cesium carbonate or under copper catalysed Ullman conditions to give the oxy bridged compounds 38, Scheme 10, which can further be elaborated to compounds of formula I as shown in Scheme 1 1 .
Scheme 10:


18, 21, 26, 27, 30, 31, 34, 37, 38
R4 = any cyclic system
R4 = -0-, -OCH2-, -(CH2)2, etc.
L is a leaving group
R1, R3, R6, R7, R8, R9, m, n, rings A and D are as defined under compound of formula I
Scheme 1 1 shows final step in the synthesis of compound of formula I.
By following similar synthetic sequences as described in Scheme 1 -1 1 , the compounds of formula I as depicted below could also be synthesized.

Ring substitutions viz. R1 , R2, R6 and R7 may be incorporated or transformed from one to another by known functional group transformation techniques at any of the steps provided hereinabove during synthesis of compound of formula I (schemes 1 to 1 1 ) or they may be protected/de-protected using suitable protecting groups wherever required using the known protection de-protection techniques as provided in Greene and Wuts 'protective groups in Organic Synthesis', Wiley and sons, 1999.
The intermediates and the compounds of the present invention are obtained in pure form in a manner known per se, for example by distilling off the solvent in vacuum and re- crystallizing the residue obtained from a suitable solvent, such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations
or subjecting it to one of the purification methods, such as column chromatography (eg. flash chromatography) on a suitable support material such as alumina or silica gel using eluent such as dichloromethane, ethyl acetate, hexane, methanol, acetone and their combinations. Preparative HPLC method is also used for the purification of molecules described herein.
Salts of compound of formula I are obtained by dissolving the compound in a suitable solvent, for example in a chlorinated hydrocarbon, such as methyl chloride or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol, which was then treated with the desired acid or base as described in Berge S.M. et al. "Pharmaceutical Salts, a review article in Journal of Pharmaceutical sciences volume 66, page 1 -19 (1977)" and in handbook of pharmaceutical salts properties, selection, and use by P.H.Einrich Stahland Camille G.wermuth , Wiley- VCH (2002). The stereoisomers of the compounds of formula I, or intermediates thereof in the present invention may be prepared by stereospecific syntheses or resolution of the racemic compound using an optically active amine, acid or complex forming agent, and separating the diastereomeric salt/complex by fractional crystallization or by column chromatography. Compounds of the present invention were prepared using synthetic schemes provided below:
Compounds 1 to 18 were prepared by following the route provided in Scheme I


Scheme I
Compounds 19 to 22 were prepared by following the route provided in Scheme II

Scheme II
Compounds 23 and 24 were prepared by following the route provided in Scheme III

Scheme
Compounds 25 and 26 were prepared by following the route provided in Scheme IV

Compound 27 was prepared by following the route provided in Scheme V

Scheme V
Compound 28 to 34 were prepared by following the route provided in Scheme VI

Scheme VI
A further embodiment of the present invention includes pharmaceutical compositions comprising any single compound, a combination of two or more compounds delineated herein, or a pharmaceutically acceptable salt thereof, with a pharmaceutically acceptable carrier or excipient.
Yet a further embodiment of the present invention is a pharmaceutical composition comprising any single compound or a combination of two or more compounds delineated herein, or a pharmaceutically acceptable salt thereof, in combination with one or more agents known in the art, with a pharmaceutically acceptable carrier or excipient. It will be further appreciated that compounds of the present invention can be administered as the sole active pharmaceutical agent, or used in combination with one or more agents to treat or prevent hepatitis C infections or the symptoms associated with HCV infection.
Other agents to be administered in combination with a compound or combination of compounds of the present invention include therapies for diseases caused by HCV infection that suppresses HCV viral replication by direct or indirect mechanisms. These agents include, but not limited to, host immune modulators (for example, interferon-alpha, pegylated interferon-alpha, consensus interferon, interferon-beta, interferon-gamma, CpG oligonucleotides and the like); antiviral compounds that inhibit host cellular functions such as inosine monophosphate dehydrogenase (for example, ribavirin and the like); cytokines that modulate immune function (for example, interleukin 2, interleukin 6, and interleukin 12); a compound that enhances the development of type 1 helper T cell response; interfering RNA; anti-sense RNA; vaccines comprising HCV antigens or antigen adjuvant combinations directed against HCV; agents that interact with host cellular components to block viral protein synthesis by inhibiting the internal ribosome entry site (IRES) initiated translation step of HCV viral replication or to block viral particle maturation and release with agents targeted toward the viroporin family of membrane proteins such as, for example, HCV P7 and the like; and any agent or combination of agents that inhibit the replication of HCV by targeting other proteins of the viral genome involved in the viral replication and/or interfere with the function of other viral targets, such as inhibitors of NS3/NS4A protease, NS3 helicase, NS5B polymerase, NS4A protein and NS5A protein.
According to yet another embodiment, the pharmaceutical compositions of the present invention may further comprise inhibitor(s) of other targets in the HCV life cycle, including, but not limited to, helicase, polymerase, metalloprotease, NS4A protein, NS5A protein, and internal ribosome entry site (IRES).
Accordingly, one embodiment of the present invention is directed to a method for treating or preventing an infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment one or more agents selected from the group consisting of a host immune modulator and a second or more antiviral agents, or a combination thereof, with a therapeutically effective amount of a compound or combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof. Examples of the host immune modulator are, but not limited to, interferon-alpha, pegylated-interferon-alpha, interferon-beta, interferon-gamma, a cytokine, a vaccine, and a vaccine comprising an antigen and an adjuvant, and said second antiviral agent inhibits replication of HCV either by inhibiting host cellular functions associated with viral
replication or by targeting proteins of the viral genome. Example of the RNA-containing virus includes, but not limited to, hepatitis C virus (HCV).
A further embodiment of the present invention is directed to a method of treating or preventing infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment an agent or combination of agents that treat or alleviate symptoms of HCV infection including cirrhosis and inflammation of the liver, with a therapeutically effective amount of a compound or combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof. Example of the RNA- containing virus includes, but not limited to, hepatitis C virus (HCV). Yet another embodiment of the present invention provides a method of treating or preventing infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment one or more agents that treat patients for disease caused by hepatitis B (HBV) infection, with a therapeutically effective amount of a compound or a combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof. An agent that treats patients for disease caused by hepatitis B (HBV) infection may be for example, but not limited thereto, L-deoxythymidine, adefovir, lamivudine or tenfovir, or any combination thereof. Example of the RNA-containing virus includes, but not limited to, hepatitis C virus (HCV).
A further embodiment of the present invention provides a method of treating or preventing infection caused by an RNA-containing virus comprising co-administering to a patient in need of such treatment one or more agents that treat patients for disease caused by human immunodeficiency virus (HIV) infection, with a therapeutically effective amount of a compound or a combination of compounds of the present invention, or a pharmaceutically acceptable salt thereof. The agent that treats patients for disease caused by human immunodeficiency virus (H IV) infection may include, but is not limited thereto, ritonavir, lopinavir, indinavir, nelfmavir, saquinavir, amprenavir, atazanavir, tipranavir, TMC-1 14, fosamprenavir, zidovudine, lamivudine, didanosine, stavudine, tenofovir, zalcitabine, abacavir, efavirenz, nevirapine, delavirdine, TMC-125, L-870812, S-1360, enfuvirtide (T- 20) or T-1249, or any combination thereof. Example of the RNA-containing virus includes, but not limited to, hepatitis C virus (HCV).
It can occur that a patient may be co-infected with hepatitis C virus and one or more other viruses, including but not limited to human immunodeficiency virus (HIV), hepatitis A virus (HAV) and hepatitis B virus (HBV). Thus also contemplated is combination therapy to treat such co-infections by co-administering a compound according to the present invention with at least one of an HIV inhibitor, an HAV inhibitor and an HBV inhibitor.
In addition, the present invention provides the use of a compound or a combination of compounds of the invention, or a therapeutically acceptable salt thereof, and one or more agents selected from the group consisting of a host immune modulator and a second or more antiviral agents, or a combination thereof, to prepare a medicament for the treatment of an infection caused by an RNA-containing virus in a patient, particularly hepatitis C virus. Examples of the host immune modulators include, but are not limited to, interferon- alpha, pegylated-interferon-alpha, interferon-beta, interferon-gamma, a cytokine, a vaccine, and a vaccine comprising an antigen and an adjuvant, and said second antiviral agent inhibits replication of HCV either by inhibiting host cellular functions associated with viral replication or by targeting proteins of the viral genome.
When used in the above or other treatments, combination of compound or compounds of the present invention, together with one or more agents as defined herein above, can be employed in pure form or, where such forms exist, in pharmaceutically acceptable salt thereof. Alternatively, such combination of therapeutic agents can be administered as a pharmaceutical composition containing a therapeutically effective amount of the compound or combination of compounds of interest, or their pharmaceutically acceptable salt thereof, in combination with one or more agents as defined hereinabove, and a pharmaceutically acceptable carrier. Such pharmaceutical compositions can be used for inhibiting the replication of an RNA-containing virus, particularly Hepatitis C virus (HCV), by contacting said virus with said pharmaceutical composition. In addition, such compositions are useful for the treatment or prevention of an infection caused by an RNA-containing virus, particularly Hepatitis C virus (HCV).
Hence, a still further embodiment of the invention is directed to a method of treating or preventing infection caused by an RNA-containing virus, particularly a hepatitis C virus (HCV), comprising administering to a patient in need of such treatment a pharmaceutical composition comprising a compound or combination of compounds of the invention or a
pharmaceutically acceptable salt thereof, and one or more agents as defined hereinabove, with a pharmaceutically acceptable carrier.
When administered as a combination, the therapeutic agents can be formulated as separate compositions which are given at the same time or within a predetermined period of time, or the therapeutic agents can be given as a single unit dosage form.
Antiviral agents contemplated for use in such combination therapy include agents (compounds or biologicals) that are effective to inhibit the formation and/or replication of a virus in a mammal, including but not limited to agents that interfere with either host or viral mechanisms necessary for the formation and/or replication of a virus in a mammal. Such agents can be selected from another anti-HCV agent; an HIV inhibitor; an HAV inhibitor; and an HBV inhibitor.
Other agents to be administered in combination with a compound of the present invention include a cytochrome P450 monooxygenase inhibitor, which is expected to inhibit metabolism of the compounds of the invention. Therefore, the cytochrome P450 monooxygenase inhibitor would be in an amount effective to inhibit metabolism of the compounds of the present invention. Accordingly, the CYP inhibitor is administered in an amount such that the bioavailability of the compounds of the present invention is increased in comparison to the bioavailability in the absence of the CYP inhibitor.
The term 'room temperature' used in the specification denotes any temperature ranging between about 20 °C to about 40 °C, except and otherwise it is specifically mentioned in the specification.
The intermediates and the compounds of the present invention may obtained in pure form in a manner known per se, for example, by distilling off the solvent in vacuum and re- crystallizing the residue obtained from a suitable solvent, such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations or subjecting it to one of the purification methods, such as column chromatography (e.g., flash chromatography) on a suitable support material such as alumina or silica gel using eluent such as dichloromethane, ethyl acetate, hexane, methanol, acetone and their
combinations. Preparative HPLC method is also used for the purification of molecules described herein.
Salts of compound of formula I can be obtained by dissolving the compound in a suitable solvent, for example in a chlorinated hydrocarbon, such as methyl chloride or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol, which was then treated with the desired acid or base as described in Berge S.M. et al. "Pharmaceutical Salts, a review article in Journal of Pharmaceutical sciences volume 66, page 1 -19 (1977)" and in handbook of pharmaceutical salts properties, selection, and use by P.H.Einrich Stahland Camille G.wermuth, Wiley- VCH (2002). Lists of suitable salts can also be found in Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p. 1445, and Journal of Pharmaceutical Science, 66, 2-19 (1977). For example, they can be a salt of an alkali metal (e.g., sodium or potassium), alkaline earth metal (e.g., calcium), or ammonium of salt.
The compound of the invention or a composition thereof can potentially be administered as a pharmaceutically acceptable acid-addition, base neutralized or addition salt, formed by reaction with inorganic acids, such as hydrochloric acid, hydrobromic acid, perchloric acid, nitric acid, thiocyanic acid, sulfuric acid, and phosphoric acid, and organic acids such as formic acid, acetic acid, propionic acid, glycolic acid, lactic acid, pyruvic acid, oxalic acid, malonic acid, succinic acid, maleic acid, and fumaric acid, or by reaction with an inorganic base, such as sodium hydroxide, potassium hydroxide. The conversion to a salt is accomplished by treatment of the base compound with at least a stoichiometric amount of an appropriate acid. Typically, the free base is dissolved in an inert organic solvent such as diethyl ether, ethyl acetate, chloroform, ethanol, methanol, and the like, and the acid is added in a similar solvent. The mixture is maintained at a suitable temperature (e.g., between 0 °C and 50 °C). The resulting salt precipitates spontaneously or can be brought out of solution with a less polar solvent. The stereoisomers of the compounds of formula I of the present invention may be prepared by stereospecific syntheses or resolution of the achiral compound using an optically active amine, acid or complex forming agent, and separating the diastereomeric salt/complex by fractional crystallization or by column chromatography.
The term "prodrug" denotes a derivative of a compound, which derivative, when administered to warm-blooded animals, e.g. humans, is converted into the compound (drug). The enzymatic and/or chemical hydrolytic cleavage of the compounds of the present invention occurs in such a manner that the proven drug form (parent carboxylic acid drug) is released, and the moiety or moieties split off remain nontoxic or are metabolized so that nontoxic metabolic products are produced. For example, a carboxylic acid group can be esterified, e.g., with a methyl group or ethyl group to yield an ester. When an ester is administered to a subject, the ester is cleaved, enzymatically or non- enzymatically, reductively, oxidatively, or hydrolytically, to reveal the anionic group. An anionic group can be esterified with moieties (e.g., acyloxymethyl esters) which are cleaved to reveal an intermediate compound which subsequently decomposes to yield the active compound.
The prodrugs can be prepared in situ during the isolation and purification of the compounds, or by separately reacting the purified compound with a suitable derivatizing agent. For example, hydroxy groups can be converted into esters via treatment with a carboxylic acid in the presence of a catalyst. Examples of cleavable alcohol prodrug moieties include substituted or unsubstituted, branched or unbranched lower alkyl ester moieties, e.g., ethyl esters, di-lower alkylamino lower-alkyl esters, e.g., dimethylaminoethyl ester, acylamino lower alkyl esters, acyloxy lower alkyl esters (e.g., pivaloyloxymethyl ester), aryl esters, e.g., phenyl ester, aryl-lower alkyl esters, e.g., benzyl ester, optionally substituted, e.g., with methyl, halo, or methoxy substituents aryl and aryl- lower alkyl esters, amides, lower-alkyl amides, di-lwer alkyl amides, and hydroxy amides. Thus the present invention further provides a pharmaceutical composition, containing the compounds of the general formula (I) as defined above, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates and its co-crystals in combination with the usual pharmaceutically acceptable carriers, diluents and the like.
The pharmaceutically acceptable carrier (or excipient) is preferably one that is chemically inert to the compound of the invention and one that has no detrimental side effects or
toxicity under the conditions of use. Such pharmaceutically acceptable carriers preferably include saline (e.g., 0.9% saline), Cremophor EL (which is a derivative of castor oil and ethylene oxide available from Sigma Chemical Co., St. Louis, MO) (e.g., 5% Cremophor EL/5% ethanol/90% saline, 10% Cremophor EL/90% saline, or 50% Cremophor EL/50% ethanol), propylene glycol (e.g., 40% propylene glycol/10% ethanol/50% water), polyethylene glycol (e.g., 40% PEG 400/60% saline), and alcohol (e.g., 40% ethanol/60% water). A preferred pharmaceutical carrier is polyethylene glycol, such as PEG 400, and particularly a composition comprising 40% PEG 400 and 60% water or saline. The choice of carrier will be determined in part by the particular compound chosen, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of the pharmaceutical composition of the present invention.
The following formulations for oral, aerosol, parenteral, subcutaneous, intravenous, intraarterial, intramuscular, interperitoneal, rectal, and vaginal administration are merely exemplary and are in no way limiting.
The pharmaceutical compositions can be administered parenterally, e.g., intravenously, intraarterially, subcutaneously, intradermal^, intrathecal^, or intramuscularly. Thus, the invention provides compositions for parenteral administration that comprise a solution of the compound of the invention dissolved or suspended in an acceptable carrier suitable for parenteral administration, including aqueous and non-aqueous, isotonic sterile injection solutions.
Overall, the requirements for effective pharmaceutical carriers for parenteral compositions are well known to those of ordinary skill in the art. See Pharmaceutics and Pharmacy Practice, J.B. Lippincott Company, Philadelphia, PA, Banker and Chalmers, eds., pages 238-250 (1982), and ASHP Handbook on Injectable Drugs, Toissel, 4th ed., pages 622- 630 (1986). Such compositions include solutions containing anti-oxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives. The compound can be administered in a physiologically acceptable diluent in a pharmaceutical carrier, such as a sterile liquid or mixture of liquids, including water, saline, aqueous
dextrose and related sugar solutions, an alcohol, such as ethanol, isopropanol (for example in topical applications), or hexadecyl alcohol, glycols, such as propylene glycol or polyethylene glycol, dimethylsulfoxide, glycerol ketals, such as 2,2-dimethyl-1 ,3-dioxolane- 4-methanol, ethers, such as poly(ethyleneglycol) 400, an oil, a fatty acid, a fatty acid ester or glyceride, or an acetylated fatty acid glyceride with or without the addition of a pharmaceutically acceptable surfactant, such as a soap or a detergent, suspending agent, such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carboxymethylcellulose, or emulsifying agents and other pharmaceutical adjuvants. Oils useful in parenteral formulations include petroleum, animal, vegetable, and synthetic oils. Specific examples of oils useful in such formulations include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral oil. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts, and suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylene polypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl-3-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof. The parenteral formulations typically will contain from about 0.5% or less to about 25% or more by weight of a compound of the invention in solution. Preservatives and buffers can be used. In order to minimize or eliminate irritation at the site of injection, such compositions can contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17. The quantity of surfactant in such formulations will typically range from about 5% to about 15% by weight. Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol. The parenteral formulations
can be presented in unit-dose or multi-dose sealed containers, such as ampoules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use. Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets.
Formulations suitable for oral administration can consist of (a) liquid solutions, such as an effective amount of a compound of the invention dissolved in diluents, such as water, saline, or orange juice; (b) capsules, sachets, tablets, lozenges, and troches, each containing a pre-determined amount of the compound of the invention, as solids or granules; (c) powders; (d) suspensions in an appropriate liquid; and (e) suitable emulsions. Liquid formulations can include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent. Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and cornstarch. Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible excipients. Lozenge forms can comprise the compound ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising a compound of the invention in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the compound of the invention, such excipients as are known in the art.
A compound of the present invention, alone or in combination with other suitable components, can be made into aerosol formulations to be administered via inhalation. A compound or epimer of the invention is preferably supplied in finely divided form along with a surfactant and propellant. Typical percentages of the compounds of the invention can be about 0.01 % to about 20% by weight, preferably about 1 % to about 10% by weight. The surfactant must, of course, be nontoxic, and preferably soluble in the propellant.
Representative of such surfactants are the esters or partial esters of fatty acids containing from 6 to 22 carbon atoms, such as caproic, octanoic, lauric, palmitic, stearic, linoleic, linolenic, olesteric and oleic acids with an aliphatic polyhydric alcohol or its cyclic anhydride. Mixed esters, such as mixed or natural glycerides can be employed. The surfactant can constitute from about 0.1 % to about 20% by weight of the composition, preferably from about 0.25% to about 5%. The balance of the composition is ordinarily propellant. A carrier can also be included as desired, e.g., lecithin, for intranasal delivery. These aerosol formulations can be placed into acceptable pressurized propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like. They also can be formulated as pharmaceuticals for non-pressured preparations, such as in a nebulizer or an atomizer. Such spray formulations can be used to spray mucosa.
Additionally, the compound of the invention can be made into suppositories by mixing with a variety of bases, such as emulsifying bases or water-soluble bases. Formulations suitable for vaginal administration can be presented as pessaries, tampons, creams, gels, pastes, foams, or spray formulas containing, in addition to the compound ingredient, such carriers as are known in the art to be appropriate.
The concentration of the compound in the pharmaceutical formulations can vary, e.g., from less than about 1 % to about 10%, to as much as 20% to 50% or more by weight, and can be selected primarily by fluid volumes, and viscosities, in accordance with the particular mode of administration selected.
For example, a typical pharmaceutical composition for intravenous infusion could be made up to contain 250 ml of sterile Ringer's solution, and 100 mg of at least one compound of the invention. Actual methods for preparing parenterally administrable compounds of the invention will be known or apparent to those skilled in the art and are described in more detail in, for example, Remington's Pharmaceutical Science (17th ed., Mack Publishing Company, Easton, PA, 1985).
It will be appreciated by one of ordinary skill in the art that, in addition to the aforedescribed pharmaceutical compositions, the compound of the invention can be formulated as inclusion complexes, such as cyclodextrin inclusion complexes, or
liposomes. Liposomes can serve to target a compound of the invention to a particular tissue, such as lymphoid tissue or cancerous hepatic cells. Liposomes can also be used to increase the half-life of a compound of the invention. Many methods are available for preparing liposomes, as described in, for example, Szoka et al., Ann. Rev. Biophys. Bioeng., 9, 467 (1980) and U.S. Patents 4,235,871 , 4,501 ,728, 4,837,028, and 5,019,369.
The present invention also provides a pharmaceutical composition, containing the compounds of the general formula (I) as defined above, its tautomeric forms, its stereoisomers, its analogs, its prodrugs, its isotopes, its metabolites, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates and its co- crystals in combination with the usual pharmaceutically employed carriers, diluents and the like, and for use in any of the methods described herein.
The compounds of the invention can be administered in a dose sufficient to treat the disease, condition or disorder. Such doses are known in the art (see, for example, the Physicians' Desk Reference (2004)). The compounds can be administered using techniques such as those described in, for example, Wasserman et al., Cancer, 36, pp. 1258-1268 (1975) and Physicians' Desk Reference, 58th ed., Thomson PDR (2004). Suitable doses and dosage regimens can be determined by conventional range-finding techniques known to those of ordinary skill in the art. Generally, treatment is initiated with smaller dosages that are less than the optimum dose of the compound of the present invention. Thereafter, the dosage is increased by small increments until the optimum effect under the circumstances is reached. The present method can involve the administration of about 0.1 μg to about 50 mg of at least one compound of the invention per kg body weight of the individual. For a 70 kg patient, dosages of from about 10 μg to about 200 mg of the compound of the invention would be more commonly used, depending on a patient's physiological response. By way of example and not intending to limit the invention, the dose of the pharmaceutically active agent(s) described herein for methods of treating or preventing a disease or condition as described above can be about 0.001 to about 1 mg/kg body weight of the subject per day, for example, about 0.001 mg, 0.002 mg, 0.005 mg, 0.010 mg, 0.015
mg, 0.020 mg, 0.025 mg, 0.050 mg, 0.075 mg, 0.1 mg, 0.15 mg, 0.2 mg, 0.25 mg, 0.5 mg, 0.75 mg, or 1 mg/kg body weight per day. The dose of the pharmaceutically active agent(s) described herein for the described methods can be about 1 to about 1000 mg/kg body weight of the subject being treated per day, for example, about 1 mg, 2 mg, 5 mg, 10 mg, 15 mg, 0.020 mg, 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 500 mg, 750 mg, or 1000 mg/kg body weight per day.
In accordance with embodiments, the present invention provides methods of treating, preventing, ameliorating, and/or inhibiting a hepatitis C virus infection comprising administering a compound of formula (I) or a salt thereof.
The compounds of the present invention are effective against the HCV 1 b genotype. It should also be understood that the compounds of the present invention can inhibit multiple genotypes of HCV. Hence, in one of the embodiment compound of the present invention are active against the 1 a, 1 b, 2a, 2b, 3a, 4a, and 5a genotypes.
The terms "treat," "prevent," "ameliorate," and "inhibit," as well as words stemming therefrom, as used herein, do not necessarily imply 100% or complete treatment, prevention, amelioration, or inhibition. Rather, there are varying degrees of treatment, prevention, amelioration, and inhibition of which one of ordinary skill in the art recognizes as having a potential benefit or therapeutic effect. In this respect, the inventive methods can provide any amount of any level of treatment, prevention, amelioration, or inhibition of the disorder in a mammal. For example, a disorder, including symptoms or conditions thereof, may be reduced by, for example, 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, or 10%. Furthermore, the treatment, prevention, amelioration, or inhibition provided by the inventive method can include treatment, prevention, amelioration, or inhibition of one or more conditions or symptoms of the disorder, e.g., cancer. Also, for purposes herein, "treatment," "prevention," "amelioration," or "inhibition" can encompass delaying the onset of the disorder, or a symptom or condition thereof.
In accordance with the invention, the term subject includes an "animal" which in turn includes a mammal such as, without limitation, the order Rodentia, such as mice, and the order Lagomorpha, such as rabbits. It is preferred that the mammals are from the order
Carnivora, including Felines (cats) and Canines (dogs). It is more preferred that the mammals are from the order Artiodactyla, including Bovines (cows) and Swine (pigs) or of the order Perssodactyla, including Equines (horses). It is most preferred that the mammals are of the order Primates, Ceboids, or Simoids (monkeys) or of the order Anthropoids (humans and apes). An especially preferred mammal is the human.
The term "viral infection" refers to the introduction of a virus into cells or tissues, e.g., hepatitis C virus (HCV). In general, the introduction of a virus is also associated with replication. Viral infection may be determined by measuring virus antibody titer in samples of a biological fluid, such as blood, using, e.g., enzyme immunoassay. Other suitable diagnostic methods include molecular based techniques, such as RT-PCR, direct hybrid capture assay, nucleic acid sequence based amplification, and the like. A virus may infect an organ, e.g., liver, and cause disease, e.g., hepatitis, cirrhosis, chronic liver disease and hepatocellular carcinoma. The term "immune modulator" refers to any substance meant to alter the working of the humoral or cellular immune system of a subject. Such immune modulators include inhibitors of mast cell-mediated inflammation, interferons, interleukins, prostaglandins, steroids, cortico-steroids, colony-stimulating factors, chemotactic factors, etc.
Following are the abbreviations used and meaning thereof in the specification: ACN: Acetonitrile. Boc: fert-butyloxycarbonyl. CDCI3: Deuterochloroform. DCM: dichloromethane. DDQ: 2,3-Dichloro-5,6-dicyano-p-benzoquinone. DIPEA: N, N- diisopropylethylamine. DME: 1 , 2-dimethoxyethane. DMF: N, N-dimethylformamide. DMSO-c6: Dimethyl sulfoxide-d6. EDCI : A/-(3-Dimethylaminopropyl)-A/'-ethylcarbodiimide. EtOAc: Ethyl acetate. HATU: 0-(7-Azabenzotriazol-1 -yl)-A/,/\/,/\/',/\/'-tetramethyluronium hexafluorophosphate. HCI: hydrochloric acid. HOBt: 1 -Hydroxybenzotriazole hydrate. HPLC - High Performance Liquid Chromatography. CD3OD: Methanol-d4. Na2S04: sodium sulphate. NaHC03: sodium bicarbonate. Pd(PPh3)4:
Tetrakis(triphenylphosphine)palladium(0). Pd-C: Palladium on carbon. PdCI2(dppf): [1 ,1 '- Bis(diphenylphosphino)ferrocene]dichloropalladium(ll). PdCl2(dppf)-CH2CI2: [1 ,1 '- Bis(diphenylphosphino)ferrocene]dichloropalladium(ll), complex with dichloromethane. pet-ether: petroleum ether. TFA: trifluoroacetic acid. THF: Tetrahydrofuran. TMS:
Trimethylsilyl. MeOH: Methanol. DMF: Dimethyl formamide. RT: room temperature. TLC: Thin layer chromatography
The following examples are provided to further illustrate the present invention and therefore should not be construed in any way to limit the scope of the present invention. All 1 HNMR spectra were determined in the solvents indicated and chemical shifts are reported in δ units downfield from the internal standard tetramethylsilane (TMS) and interproton coupling constants are reported in Hertz (Hz). In case of mixture of the isomers, the peak values given are for the dominant isomer (rotamer/tautomer).
Example 1 : Synthesis of (S)-tert-butyl 2-(7-bromo-1 H-naphtho[1 ,2-d]imidazol-2- yl)pyrrolid

To a solution of (S)-tert-butyl 2-(7-bromo-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (1 .5 g, 3.59 mmol) (ref. WO2010/65688 A1 ) in toluene (50 ml) was added DDQ (1.22 g, 5.38 mmol) and the reaction mixture was refluxed for 5 h, after which all the volatiles were removed under reduced pressure and the reaction mass was extracted with ethyl acetate and washed with water. The ethyl acetate layer was washed with a saturated solution of NaHC03 followed by brine. The combined organic extracts were dried over Na2S04 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 30-40% ethyl acetate/Hexane) to yield a yellowish white solid (850 mg, 57%). 1 H-NMR (400 MHz, CD3OD): δ 8.05-8.03 (m, 1 H), 7.62-7.50 (m, 4H), 5.24-5.22 (m, 1 H), 3.61 -3.50 (m, 1 H), 3.15-3.10 (m, 1 H), 2.29-2.19 (m, 2H), 2.18-2.01 (m, 2H), 1 .54-1 .34 (m, 9H); LCMS: m/z = 416.1 (M+H)+
Example 2: Synthesis of (S)-tert-butyl 2-(7-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1-carboxylate:

15 mL of dioxane was degassed by passing nitrogen gas for 10 min. in a microwave vial, and then 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1 ,3,2-dioxaborolane) (488 mg, 1 .92 mmol), (S)-tert-butyl 2-(7-bromo-3H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (400 mg, 0.96 mmol), potassium acetate (377 mg, 3.84 mmol) and PdCI2(dppf)-CH2Cl2 adduct (78 mg, 0.10 mmol) were added. The vial was sealed and irradiated under microwave at 1 15 °C for 45 min. After cooling, the reaction mixture was added to water and the reaction mass was extracted with 20 % CHCI3-MeOH and dried over Na2S04 and concentrated. Purification was done using Combiflash™ by eluting with 2-5 % MeOH in DCM to yield the title compound as a white foam (300 mg, 68%). 1 H-NMR (400 MHz, CDCI3): δ 8.46 (s, 1 H), 7.97-7.95 (m, 1 H), 7.71 -7.47 (m, 3H), 5.23-5.22 (brs, 1 H), 3.48-3.41 (m, 1 H), 3.23-3.20 (m, 1 H), 2.26-2.19 (m, 2H), 2.61 -2.41 (m, 2H), 1 .41 (s, 9H), 1 .30-1 .22 (m, 12H); LCMS: m/z = 464.3 (M+H)+
Example 3: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1-(6-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide (Compound 1)
Step 1 : tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (1 a)
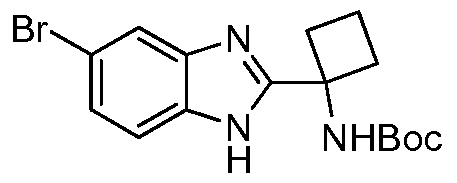
To a solution of 1 -((tert-butoxycarbonyl)amino)cyclobutanecarboxylic acid (0.75 g, 3.48 mmol) in 7 mL DMF, Hunig's base (1 .52 mL, 8.71 mmol) was added followed by EDCI.HCI (0.87 g, 4.53 mmol). To this stirred mixture at room temperature, HOBt was added (0.53 g, 3.48 mmol) after 10 min. followed by 4-bromobenzene-1 ,2-diamine (0.67 g, 3.59 mmol) after another 10 min. The brown mixture was allowed to stir overnight, after which ice cold
water was added and the reaction mixture was extracted with 100 mL ethyl acetate twice. The combined ethyl acetate extracts were dried over Na2S04 and concentrated in the rotavap to obtain a brown foam containing a mixture of tert-butyl (1 -((2-amino-5- bromophenyl)carbamoyl)cyclobutyl)carbamate and tert-butyl (1 -((2-amino-4- bromophenyl)carbamoyl)cyclobutyl)carbamate as the two amide regio isomers.
The mixture of isomers as synthesized above was taken in 10 mL of acetic acid and heated at 60 °C for 4 h. The reaction was cooled and most of the acetic acid was removed in the rotavap and the remaining traces of acid was neutralised with a satd. solution of NaHC03. The compound was extracted with 100 mL ethyl acetate twice. The combined ethyl acetate extracts were dried over Na2S04 and concentrated in the rotavap to obtain a brown mass which was purified by column chromatography (silica gel; 20-30% EtOAc/hexane) to yield a pale brown coloured solid (0.85 g, 69%). 1 H-NMR (400 MHz, CDCI3), 6 10.62 (brs, 1 H), 7.91 -7.88 (m, 0.5H), 7.64-7.59 (m, 1 H), 7.36-7.29 (m, 1 .5H), 5.32 (s, 1 H), 4.72 (brs, 1 H), 2.96-2.93 (m, 2H), 2.45-2.38 (m, 2H), 2.20-2.09 (m, 2H), 1 .48 (s, 9H); LCMS: m/z = 366.2 (79Br) & 368.2 (81 Br).
Step 2: tert-butyl (1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-benzo[d]imidazol- 2-yl)cyclobutyl)carbamate (1 b)

4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1 ,3,2-dioxaborolane) (0.42 g, 1 .64 mmol) and tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (1 a) (0.3 g, 0.82 mmol) was added to 15 mL of degassed dioxane in a microwave vial followed by potassium acetate (0.24 g, 2.46 mmol) and a catalytic amount of Pd(PPh3)4 (7 mol%). The mixture was subjected to microwave at 120 °C for 45 min and the reaction mass was extracted with ethyl acetate and washed with water. The ethyl acetate extracts were dried over Na2S04 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 2% MeOH/DCM) to yield a white foam (0.18 g, 54%). 1 H-NMR (400 MHz, CDCI3), 6 8.07 (brs, 1 H), 7.75-7.66 (m, 2H), 7.51 -7.48 (m, 1 H), 5.52 (s, 1 H),
3.06-2.90 (m, 2H), 2.50-2.37 (m, 2H), 2.19-2.03 (m, 2H), 1 .37 (s, 12H), 1 .27 (s, 9H); LCMS: m/z = 414.1 (M+H)+.
Step 3: tert-butyl 2-(8-(2-(1 -((tert-butoxycarbonyl)arriino)cyclobutyl)-1 H-benzo[d]irriidazol- 5-yl)-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (1 c)

tert-butyl (1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-benzo[d]imidazol-2- yl)cyclobutyl)carbamate (1 b) (100 mg, 0.242 mmol) and tert-butyl 2-(8-bromo-4,5-dihydro- 3H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (121 mg, 0.278 mmol) [Prepared according to procedure reported in WO 2009/102633 A1] was added to a degassed mixture of DME-H20 (2:1 , 3 mL) in a microwave vial followed by Na2C03 (103 mg, 0.97 mmol) and a catalytic amount of Pd(PPh3)4 (7 mol%). The mixture was subjected to microwave at 120 °C for 30 min. and the reaction mass was extracted with ethyl acetate and washed with water. The ethyl acetate extracts were dried over Na2S04 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 2-3% MeOH/DCM) to yield a yellowish white solid (130 mg, 84%). 1 H-NMR (400 MHz, CDCI3): 6 7.71 -7.66 (m, 2H), 7.57-7.46 (m, 3H), 7.41 -7.38 (m, 1 H), 5.33-5.31 (m, 2H), 5.02-4.94 (m, 2H), 4.33-4.27 (m, 2H), 3.44-3.41 (m, 2H), 3.20-3.14 (m, 2H), 3.01 -2.99 (m, 2H), 2.47-2.44 (m, 2H), 1 .97-1 .94 (m, 2H), 1 .52-1 .48 (m, 18H) ; LCMS: m/z = 641 .3(M+H)+.
Step 4: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-((S)-1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-8-yl)- 1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 1 )

Example 4: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)- 3-methylbutanamide (Compound 2):
Step 1 : tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)carbamate (2a)

1 ,4-bis(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzene (0.49 g, 1 .47 mmol) and tert- butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (0.18 g, 0.49 mmol) was added to a degassed mixture of DME-H20 (2:1 , 6 mL) in a microwave vial followed by
Na2C03 (0.31 g, 2.95 mmol) and a catalytic amount of Pd(PPh3)4 (7 mol%). The mixture was subjected to microwave at 1 15 °C for 45 min and the reaction mass was extracted with ethyl acetate and washed with water. The ethyl acetate extracts were dried over Na2S04 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 20-30% EtOAc/hexane) to yield an off white foam (0.20 g, 85%). 1 H-NMR (400 MHz, CDCI3): 6 10.59 (brs, 1 H), 7.91 (d, J = 8Hz, 2H), 7.82 (s, 1 H), 7.67 (d, J = 8Hz, 2H),7.57-7.47 (m, 2H), 5.32 (s, 1 H), 3.03-2.99 (m, 2H), 2.47-2.41 (m, 2H), 2.16-2.08 (m, 2H), 1 .48 (s, 12H), 1 .36 (s, 9H); LCMS: m/z = 490.4(M+H)+. Step 2: tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-5-yl)phenyl)-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (2b)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (2a) and (S)-tert-butyl 2-(8-bromo-4,5- dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 -carboxylate [Prepared according to procedure reported in WO 2009/102633 A1 ] by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3), 6 10.62 (brs, 1 H), 7.74-7.65 (m, 6H), 7.58-7.53 (m, 2H), 7.50-7.45 (m, 2H), 5.03-4.95 (m, 2H), 4.38-4.32 (m, 2H), 3.44-3.41 (m, 2H), 3.24-3.15 (m, 4H), 3.08-2.98 (m 2H), 2.47-2.42 (m, 2H), 2.22-2.1 1 (m, 2H), 1 .98-1 .89 (m, 2H), 1 .51 -1 .49 (m, 18H); LCMS: m/z = 717.1 (M+H)+.
Step 3. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol- 8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 2)

Example 5: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide (Compound 3):
Step 1 : tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-5-yl)phenyl)-4,5-dihydro-3H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (3a)
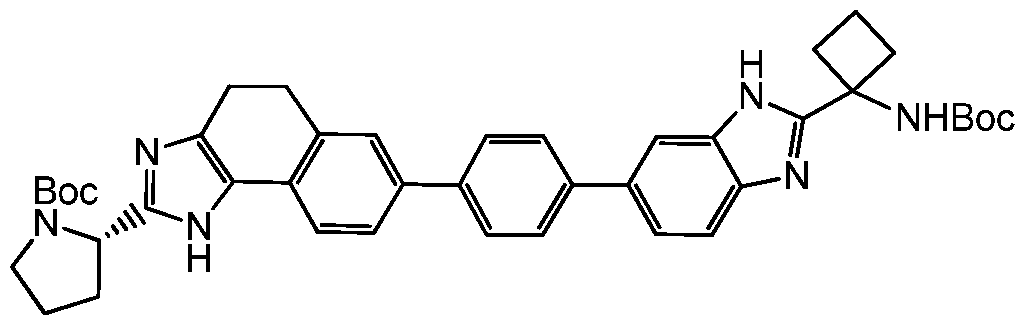
Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (2a) and (S)-tert-butyl 2-(7-bromo-4,5- dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate [Prepared according to procedure reported in WO 2009/102633 A1] by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3), 6 10.84 (brs, 1 H), 10.60 (brs, 1 H), 7.85-7.67 (m, 6H), 7.57-7.48 (m, 4H), 5.38-5.36 (m, 1 H), 5.03-4.99 (m, 1 H), 3.46-3.42 (m, 2H), 3.13-3.09 (m, 2H), 3.04-3.01 (m, 2H), 2.95-2.90 (m, 2H), 2.48-2.42 (m, 2H), 2.14- 2.06 (m, 4H), 1 .98-1 .85 (m, 2H), 1 .53-1 .49 (m, 18H); LCMS: m/z = 701 .2(M+H)+.
Step 2. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imi
yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 3)

Synthesized from tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-5-yl)phenyl)-4,5-dihydro-3H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (3a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 7.96-7.90 (m, 3H), 7.83-7.62 (m, 4H), 7.58-7.52 (m, 3H), 5.16-5.13 (m, 2H), 4.55-4.23 (m, 2H), 4.04-4.01 (m, 2H), 3.95-3.88 (m, 2H), 3.74 (s, 3H), 3.67 (s, 3H), 2.85-2.79 (m, 2H), 2.63-2.55 (m, 2H), 2.38-2.27 (m, 2H), 2.22-2.15 (m, 2H), 2.1 1 -2.04 (m, 2H), 1 .05-0.90 (m, 12H); LCMS: m/z = 815.1 (M+H)+.
Example 6: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclobutyl)-3-methylbutanamide (Compound 4):
Step 1 : (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (4a)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (2a) and (S)-tert-butyl 2-(8-bromo-1 ,4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate [Prepared according to procedure reported in WO 2009/102633 A1 ] by following an analogous
procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.79-7.68 (m, 2H), 7.74 (s, 4H), 7.58-7.46 (m, 4H), 3.56-3.49 (m, 1 H), 3.19-3.08 (m, 2H), 2.81 -2.62 (m, 4H), 2.49-2.38 (m, 4H), 2.07-1 .92 (m, 4H), 1 .69-1 .49 (m, 4H), 1 .25(s, 18H); LCMS: m/z = 715.5 (M+H)+.
Step 2. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2- d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide
(Compound 4)

Synthesized from (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (4a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.79-7.68 (m, 2H), 7.74 (s, 4H), 7.58-7.49 (m, 3H), 7.34 (s, 1 H), 5.18- 5.06 (m, 1 H), 4.28-4.21 (m, 2H), 3.98-3.91 (m, 2H), 3.74 (s, 3H), 3.67 (s, 3H), 2.99-2.89 (m, 2H), 2.88-2.83 (m, 2H), 2.61 -2.58 (m, 4H), 2.39-2.37 (m, 2H), 2.26-2.06 (m, 6H), 1 .69-1 .49 (m, 4H), 1 .09-1 .04 (m, 12H); LCMS: m/z = 829.5 (M+H)+.
Example 7: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 5):
Step 1 : Synthesis of (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)- 1 H-benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (5a)
NHBoc
Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (2a) and (S)-tert-butyl 2-(7-bromo-1 H- naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (prepared as per Example 1 ) by following an analogous procedure described in Step 3, Example 3. LCMS: m/z = 699.4 (M+H)+.
Step 2. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 5)

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (5a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 8.64-8.43 (m, 1 H), 8.34-8.26 (m, 1 H), 7.94-7.92 (m, 1 H), 7.87-7.85 (m, 2H), 7.81 -7.76 (m, 3H), 7.69-7.61 (m, 3H), 4.89-4.30 (m, 2H), 4.29-4.02 (m, 2H), 4.00-3.78 (m, 3H), 3.69-3.65 (m, 1 H), 3.78 (s, 3H), 3.66 (s, 3H), 2.96-2.94 (m, 2H), 2.68-2.66 (m, 2H), 2.39-2.32 (m, 2H), 2.16-1 .98 (m, 5H), 1 .08-0.92 (m, 12H); LCMS: m/z = 813.5(M+H)+.
Example 8: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopentyl)-3-methylbutanamide (Compound 6):
Step 1 : tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6a)

Synthesized from 1 -((tert-butoxycarbonyl)amino)cyclopentanecarboxylic acid and 4- bromobenzene-1 ,2-diamine by following an analogous procedure described in Step 1 , Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.90-7.85 (m, 1 H), 7.64-7.52 (m, 1 H),7.48-7.47 (m,1 H), 5.75 (brs, 1 H), 2.52-2.41 (m, 2H), 2.31 -2.28 (m, 2H), 1 .88-1 .85 (m, 4H), 1 .46 (s, 9H); LCMS: m/z = 380.1 (79Br) & 382.1 (81 Br).
Step 2: tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6b)

Synthesized from tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6a) by following an analogous procedure described in Step 1 , Example 4. 1H-NMR (400 MHz, CDCI3): 6 7.91 -7.89 (d, J = 8Hz, 2H), 7.81 (s, 1 H), 7.67-7.65 (d, J = 8Hz, 2H), 7.54- 7.51 (m, 2H), 5.07 (brs, 1 H), 2.60-2.57 (m, 2H), 2.40-2.31 (m, 2H), 1 .90-1 .88 (m, 4H), 1 .51 -1 .46 (m, 12H), 1 .38-1 .36 (s, 9H); LCMS: m/z = 504.3 (M+H)+.
Step 3: Synthesis of (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)- 1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (6c)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6b) and (S)-tert-butyl 2-(8-bromo-4,5- dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 8.03-
7.01 (m, 10H), 5.03-4.97 (m, 1 H), 4.37-4.30 (m, 1 H), 3.53-3.10 (m, 4H), 2.58-1 .89 (m, 14H), 1 .54-1 .47 (m, 18H); LCMS: m/z =731 .4.
Step 4: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]i
8-yl)phenyl)-1 -benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 6)

Synthesized from (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (6c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.91 -7.20 (m, 12H), 5.41 -5.03 (m, 1 H), 4.24-4.23 (m, 1 H), 3.94-3.92 (m, 2H), 3.75 (s, 3H), 3.72 (s, 3H), 3.52-3.49 (m, 1 H), 3.18-3.10 (m, 2H), 2.67-2.57 (m, 2H), 2.31 -1 .92 (m, 13H), 1 .39-1 .36 (m, 2H), 1 .01 -0.89 (m, 12H); LCMS: m/z = 845.4 (M+H)+.
Example 9: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopentyl)-3-methylbutanamide (Compound 7):
Step 1 : (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (7a)

Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2- d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide
(Compound 7)

Synthesized from (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (7a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.84-7.47 (m, 10H), 5.24-5.18 (m, 1 H), 4.63-4.59 (m, 1 H), 3.93-3.92 (m, 2H), 3.75 (s, 3H), 3.69 (s, 3H), 2.96-2.94 (m, 6H), 2.67-2.57 (m, 2H), 2.29-1 .94 (m, 14H), 1 .01 -0.92 (m, 12H); LCMS: m/z = 843.4 (M+H)+.
Example 10: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide (Compund 8):
Step 1 : (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (8a)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6b) and (S)-tert-butyl 2-(7-bromo-4,5- dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 7.75- 7.69 (m, 5H), 7.57-7.48 (m, 5H), 5.07-5.02 (m, 2H), 3.50-3.46 (m, 2H), 3.12-3.05 (m, 2H), 2.92-2.89 (m, 2H), 2.61 -2.58 (m, 2H), 2.35-2.32 (m, 2H), 2.22-2.17 (m, 2H), 2.06-1 .99 (m, 2H), 1 .91 -1 .89 (m, 4H), 1 .53-1 .47 (m, 18H); LCMS: m/z =715.4
Step 2. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-7- yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 8)

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (8a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 7.73-7.67 (m, 5H), 7.55-7.51 (m, 5H), 5.15-5.13 (m, 1 H), 4.63-4.60 (m, 1 H), 4.25-4.23 (m, 1 H), 3.95-3.88 (m, 2H), 3.75 (s, 3H), 3.66 (s, 3H), 3.49-3.47 (m, 2H), 3.14-3.07 (m, 3H), 2.85-2.82 (m, 2H), 2.59-2.52 (m, 2H), 2.38-1 .90 (m, 8H), 1 .01 -0.90 (m, 14H); LCMS: m/z = 829.4 (M+H)+.
Example 11 : Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 9):
Step 1 : (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (9a).

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6b) and (S)-tert-butyl 2-(7-bromo-1 H- naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (obtained as per Example 1 ) by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 8.19-7.48 (m, 12H), 5.29-5.26 (m, 1 H), 5.12-5.09 (m, 1 H), 3.56-3.50 (m, 2H), 3.26-3.15 (m, 1 H), 2.62-2.59 (m, 2H), 2.32-2.29 (m, 2H), 2.08-2.06 (m, 2H), 1 .95-1 .89 (m, 4H), 1 .62 (s, 9H), 1 .47 (s, 9H); LCMS: m/z =713.4
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 9)

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (9a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 8.30-8.26 (m,
2H), 7.96-7.76 (m, 6H), 7.70-7.58 (m, 4H), 5.41 -5.36 (m, 1 H), 4.33-4.31 (m, 1 H), 4.17-3.92 (m, 2H), 3.75 (s, 3H), 3.67 (s, 3H), 2.64-2.49 (m, 4H), 2.30-1 .91 (m, 10H), 1 .01 -0.1 1 (m, 14H); LCMS: m/z = 827.4 (M+H)+.
Example 12: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopropyl)-3-methylbutanamide (Compound 10):
Step 1 : tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclopropyl)carbamate (10a)

Synthesized from 1 -((tert-butoxycarbonyl)amino)cyclopropanecarboxylic acid and 4- bromobenzene-1 ,2-diamine by following an analogous procedure described in Step 1 , Example 3. LCMS: m/z = 352.3 (79Br) & 354.3 (81 Br) (M+H)+.
Step 2: tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopropyl)carbamate (10b)

Synthesized from tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclopropyl)carbamate (10a) by following an analogous procedure described in Step 1 , Example 4. 1 H-NMR (400 MHz, CDCI3): 6 10.50 (brs, 1 H), 7.89 (d, J = 8Hz, 2H), 7.83 (s, 1 H), 7.65 (d, J = 8Hz, 2H), 7.57-7.47 (m, 2H), 5.30 (s, 1 H), 0.98-0.97 (m, 2H), 0.55-0.52 (m, 2H), 1 .48 (s, 9H), 1 .36 (s, 12H); LCMS: m/z = 476.1 (M+H)+.
Step 3: (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopropyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (10c)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclopropyl)carbamate (10b) and (S)-tert-butyl 2-(8-bromo-4,5- dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 10.35 (brs, 1 H), 7.74-7.62 (m, 4H), 7.55-7.50 (m, 2H), 7.49-7.38 (m, 2H), 5.02-4.98 (m, 2H), 4.38-4.32 (m, 2H), 3.49-3.44 (m, 2H), 3.26-3.124 (m, 2H), 3.08-2.99 (m, 2H), 2.22-2.10 (m, 2H), 1 .98-1 .90 (m, 2H), 1 .69-1 .67 (m, 2H), 1 .49-1 .45 (m, 18H), 1 .31 -1 .29 (m, 2H); LCMS: m/z =703.4.
Step 4: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol- 8-yl)phenyl)-1 -benzo[d]imidazol-2-yl)cyclopropyl)-3-methylbutanamide (Compound 10)

Synthesized from (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopropyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (10c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.75-7.69 (m, 6H), 7.55-7.53 (m, 2H), 7.42 (d, J = 8Hz, 2H), 7.35-7.31 (brs, 2H), 5.17-5.10 (m, 2H), 4.35-4.24 (m, 2H), 3.95-3.92 (m, 2H), 3.88 (s, 3H), 3.66 (s, 3H), 3.51 -3.49 (m, 2H), 3.17-3.12 (m, 2H), 3.05-2.98 (m, 2H), 2.86-2.80 (m, 2H), 2.63-2.56 (m, 2H), 2.23-2.16 (m, 2H), 2.10-2.02 (m, 2H), 1 .05-0.92 (m, 12H); LCMS: m/z = 817.4 (M+H)+.
Example 13: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopropyl)-3-methylbutanamide (Compound 11):
Step 1 : (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopropyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (1 1 a)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclopropyl)carbamate (10b) and (S)-tert-butyl 2-(8-bromo- 1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate
[Prepared according to procedure reported in WO 2009/102633 A1 ] by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 10.83 (brs, 1 H), 7.74-7.62 (m, 4H), 7.55-7.50 (m, 4H), 7.49-7.38 (m, 2H), 5.02-4.98 (m, 2H), 3.49-3.44 (m, 2H), 3.26-3.12 (m, 2H), 3.08-2.98 (m 2H), 2.47-2.42 (m, 2H), 2.22-2.1 1 (m, 2H), 1 .98-1 .89 (m, 2H), 1 .51 -1 .49 (m, 18H), 1 .31 -1 .28 (m, 2H); LCMS: m/z =701 .5
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2- d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopropyl)-3-methylbutanamide
(Compound 1 1

Synthesized from (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopropyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (1 1 a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic
acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.73-7.70 (m, 2H), 7.56-7.52 (m, 4H), 7.58-7.49 (m, 4H), 5.18-5.06 (m, 1 H), 4.28-4.21 (m, 1 H), 3.94-3.92 (m, 2H), 3.67 (s, 3H), 3.50 (s, 3H), 2.99-2.89 (m, 4H), 2.88-2.83 (m, 2H), 2.61 -2.58 (m, 4H), 2.39-2.37 (m, 2H), 2.26-2.06 (m, 6H), 1 .69-1 .49 (m, 4H), 1 .03-0.92 (m, 12H); LCMS: m/z = 815.5 (M+H)+.
Example 14: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)- 3-methylbutanamide (Compound 12):
Step 1 : tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12a)

Synthesized from 1 -((tert-butoxycarbonyl)amino)cyclohexanecarboxylic acid and 4- bromobenzene-1 ,2-diamine by following an analogous procedure described in Step 1 , Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.72 (s, 1 H), 7.45 (d, J = 8Hz, 1 H), 7.32 (dd, J = 1 .6Hz, 8Hz, 1 H), 4.93 (brs, 1 H), 2.48-2.31 (m, 2H), 2.19-2.12 (m, 2H), 1 .74-1 .66 (m, 4H), 1 .62-1 .53 (m, 2H), 1 .48-1 .37 (m, 9H); LCMS: m/z = 394.1 (79Br) & 396.1 (81 Br) (M+H)+.
Step 2: tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H-benzo[d] imidazol-2-yl)cyclohexyl)carbamate (12b)
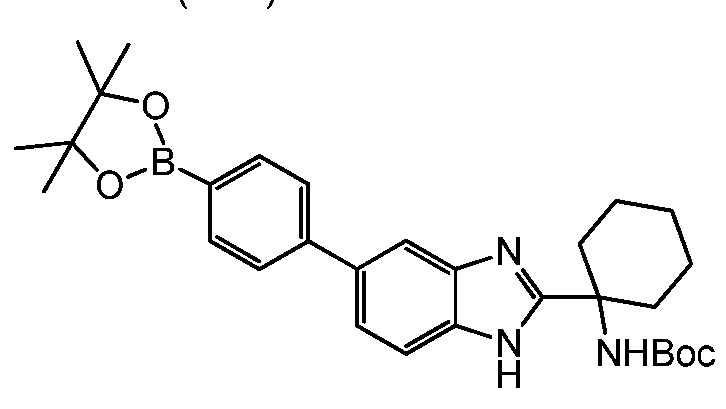
Synthesized from tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12a) and 1 ,4-bis(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)benzene by following an analogous procedure described in Step 1 , Example 4. 1 H-NMR (400 MHz, CDCI3): δ 10.90 (brs, 1 H), 7.91 (d, J = 8Hz, 2H), 7.73 (s, 1 H), 7.66 (d, J = 8Hz, 2H), 7.5-7.51 (m, 2H), 5.91
(brs, 1 H), 2.44-2.40 (m, 2H), 2.25-2.22 (m, 2H), 1 .75-1 .65 (m, 4H), 1 .64-1 .59 (m, 2H), 1 .38 (s, 9H), 1 .25 (s, 12H); LCMS: m/z = 518.1 (M+H)+.
Step 3: (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (12c)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12b) and (S)-tert-butyl 2-(8-bromo-4,5- dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 7.75- 7.43 (m, 10H), 5.43-5.41 (m, 1 H), 4.92-4.90 (m, 2H), 4.38-4.36 (m, 2H), 3.55-3.44 (m, 2H), 3.27-3.25 (m, 2H), 2.51 -1 .89 (m, 12H), 1 .71 -1 .66 (m, 2H), 1 .48-1 .25 (m, 18H); LCMS: m/z = 745.5 (M+H)+.
Step 5. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol- 8-yl)phenyl)-1 -benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 12)

Synthesized from (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (12c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.75-7.64 (m, 4H), 7.59-7.55 (m, 2H), 7.45-7.41 (m, 2H), 7.38-7.35 (m, 2H), 5.17-5.13 (m, 1 H), 4.35-4.30 (m, 2H), 4.02-3.99 (m, 2H), 3.96-3.92 (m, 2H), 3.74 (s,
3H), 3.66 (s, 3H), 3.17-3.14 (m, 4H), 2.77-2.65 (m, 4H), 2.33-1 .99 (m, 8H), 1 .75-1 .64 (m, 4H), 1 .06-0.88 (m, 12H); LCMS: m/z = 859.4 (M+H)+.
Example 15: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide (Compound 13):
Step 1 : (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (13a)

Synthesized from tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)- 1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12b) and (S)-tert-butyl 2-(8-bromo- 1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): 6 10.84 (brs, 1 H), 7.89-7.83 (m, 4H), 7.62-7.49 (m, 4H), 7.44-7.39 (m, 2H), 5.02- 4.98 (m, 2H), 3.49-3.44 (m, 2H), 3.26-3.12 (m, 2H), 3.08-2.98 (m 4H), 2.47-2.42 (m, 2H), 2.22-2.1 1 (m, 4H), 1 .98-1 .89 (m, 4H), 1 .72-1 .64 (m, 6H), 1 .51 -1 .49 (m, 18H); LCMS: m/z =743.4
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2- d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide
(Compound 13)

Synthesized from (S)-tert-butyl 2-(8-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 ,4,5,6-tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (13a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 8.30-8.26 (br m, 2H), 7.80-7.78 (m, 2H), 7.75-7.72 (m, 4H), 7.64-7.52 (m, 4H), 5.21 -5.18 (m, 1 H), 4.26-4.24 (m, 1 H), 4.17-3.92 (m, 2H), 3.91 -3.88 (m, 2H), 3.73 (s, 3H), 3.67 (s, 3H), 3.02-2.72 (m, 4H), 2.63-2.50 (m, 4H), 2.30-1 .91 (m, 10H), 1 .78-1 .66 (m, 4H), 1 .01 -0.90 (m, 12H); LCMS: m/z = 857.4 (M+H)+.
Example 16: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 14):
Step 1 : (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl -1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (14a)
Synthesized from (S)-tert-butyl 2-(7-bromo-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (obtained as per example 1 ) and tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12b) by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.88-7.48 (m, 12H), 5.26-5.24 (m, 1 H), 5.02-4.97 (m, 1 H), 3.56-3.41 (m, 2H), 3.20-3.10 (m, 2H), 2.50-2.40 (m, 2H), 2.26-2.18 (m, 4H), 2.01 -1 .90 (m, 2H) 1 .85-1 .72 (m, 4H), 1 .56-1 .48 (m, 18H); LCMS: m/z =727.4.
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 14):

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (14a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 8.30-8.27 (m, 2H), 7.96-7.94 (m, 2H), 7.89-7.87 (m, 2H), 7.80-7.77 (m, 4H), 7.60-7.57 (m, 2H), 5.39-5.36 (m, 2H), 4.30-4.29 (m, 2H), 4.09-4.01 (m, 4H), 3.75 (s, 3H), 3.67 (s, 3H), 2.73-2.67 (m, 1 H), 2.51 -2.48 (m, 1 H), 2.30-2.14 (m, 2H), 2.07-2.02 (m, 2H) 1 .80-1 .73 (m, 4H), 1 .51 -1 .40 (m, 2H), 1 .33-1 .31 (m, 2H), 1 .04-0.89 (m, 12H); LCMS: m/z = 841 .4 (M+H)+.
Example 17: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 15):
Step 1 : (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (15a)

Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-7- yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 15)

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (15a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 7.78-7.67 (m, 5H), 7.59-7.54 (m, 5H), 5.15-5.13 (m, 1 H), 4.66-4.62 (m, 2H), 4.27-4.21 (m, 2H), 3.95-3.88 (m, 2H), 3.78 (s, 3H), 3.67 (s, 3H), 3.49-3.46 (m, 2H), 3.12-3.08 (m, 3H), 2.85-2.82 (m, 2H), 2.59-2.52 (m, 2H), 2.38-1 .90 (m, 8H), 1 .78-1 .69 (m, 2H), 1 .01 -0.88 (m, 12H); LCMS: m/z = 843.5 (M+H)+.
Example 18: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N-ethyl-3- methylbutanamide (Compound 16):
Step 1 : tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)(ethyl)carbamate (16a):

Synthesized from 1 -((tert-butoxycarbonyl)(ethyl)amino)cyclobutanecarboxylic acid [Synthesized according to procedure reported in WO 2010002655 A2] and 4- bromobenzene-1 ,2-diamine by following an analogous procedure described in Step 1 , Example 3. 1 H-NMR(400 MHz, CDCI3): 6 10.49 (brs, 1 H), 7.93 (s, 1 H), 7.66-7.64 (m, 1 H), 7.37-7.35 (d, J = 7.2Hz, 1 H), 3.19-3.14 (m, 2H), 3.00-2.80 (m, 2H), 2.57-2.49 (m, 2H), 1 .87-1 .79 (m, 2H), 1.51 (s, 9H), 0.94-0.90 (m, 3H); LCMS: m/z = 394.1 (79Br) & 396.1 (81 Br) (M+H)+.
Step 2: tert-butyl ethyl(1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)carbamate (16b):

Synthesized from tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2- yl)cyclobutyl)(ethyl)carbamate (16a) by following an analogous procedure described in Step 1 , Example 4. 1 H-NMR (400 MHz, CDCI3): 6 10.59 (brs, 1 H), 7.89 (d, J = 8Hz, 2H), 7.86 (s, 1 H), 7.65 (d, J = 8Hz, 2H),7.54-7.46 (m, 2H), 5.30 (s, 1 H), 3.03-2.99 (m, 2H), 2.57- 2.49 (m, 2H), 2.47-2.41 (m, 2H), 2.16-2.08 (m, 2H), 1.48 (s, 12H), 1 .36 (s, 9H), 0.94-0.91 (m, 3H); LCMS: m/z = 518.3 (M+H)+.
Step 3: (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)(ethyl)amino)cyclobutyl)-1 H-benzo [d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (16c):

Step 5. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-7- yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N-ethyl-3-methylbutanamide (Compound

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)(ethyl)amino)cyclobutyl)- 1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine- 1 -carboxylate (16c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H NMR (400 MHz, CDCI3): δ 7.85 (s, 1 H), 7.75-7.73 (m, 5H), 7.70-7.65 (m, 2H), 7.60-7.54 (m, 4H), 5.20 (m, 1 H), 4.35-4.33 (m, 1 H), 4.23-4.20 (m, 1 H), 3.60 (s, 3H), 3.35 (s, 3H), 3.20-3.00 (m, 4H), 2.95-2.80 (m, 4H), 2.55 (m, 2H), 2.40-2.10 (m, 4H), 2.12-1 .90 (m, 6H), 1 .10-0.99 (m, 3H), 0.95-0.93 (m, 12H); LCMS: m/z = 843.4 (M+H)+.
Example 19: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N,3- dimethylbutanamide (Compound 17):
Step 1 : tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclobutyl)(methyl)carbamate (17a).
 Synthesized from 1 -((tert-butoxycarbonyl)(methyl)amino)cyclobutanecarboxylic acid [Synthesized according to procedure reported in WO 2010002655 A2] and 4- bromobenzene-1 ,2-diamine by following an analogous procedure described in Step 1 , Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.66-7.58 (m, 1 H), 7.58-7.31 (m, 2H), 2.93-2.91 (m, 2H), 2.57 (s, 3H), 2.55-2.49 (m, 2H), 1 .83-1 .81 (m, 2H), 1 .44 (s, 9H); LCMS: m/z = 379.9 (79Br) & 381 .9 (81 Br) (M+H)+.
Synthesized from 1 -((tert-butoxycarbonyl)(methyl)amino)cyclobutanecarboxylic acid [Synthesized according to procedure reported in WO 2010002655 A2] and 4- bromobenzene-1 ,2-diamine by following an analogous procedure described in Step 1 , Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.66-7.58 (m, 1 H), 7.58-7.31 (m, 2H), 2.93-2.91 (m, 2H), 2.57 (s, 3H), 2.55-2.49 (m, 2H), 1 .83-1 .81 (m, 2H), 1 .44 (s, 9H); LCMS: m/z = 379.9 (79Br) & 381 .9 (81 Br) (M+H)+.
Step 2: tert-butyl methyl(1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)carbamate (17b)

Synthesized from tert-butyl (1 -(5-bromo-1 H-benzo[d]imidazol-2- yl)cyclobutyl)(methyl)carbamate (17a) by following an analogous procedure described in Step 1 , Example 4. LCMS: m/z = 503.7 (M+H)+.
Step 3: (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)(methyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (17c)

Synthesized from tert-butyl methyl(1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (17b) and (S)-tert-butyl 2-(7- bromo-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 7.89-7.82 (m, 1 H), 7.55 (m, 3H), 7.68-7.65 (m, 2H), 7.59-7.54 (m, 4H), 4.61 (m, 1 H), 4.13- 4.10 (m, 1 H), 3.70 (m, 1 H), 3.50-3.49 (m, 1 H), 3.16-3.08 (m, 4H), 2.89-2.85 (m, 2H), 2.75- 2.70 (m, 4H), 2.41 (m, 1 H), 2.07-2.05 (m, 2H), 2.02 (m, 1 H), 1 .98-1 .97 (m, 2H), 1 .49-1 .44 (m, 4H), 1 .38-1 .21 (m, 14H); LCMS: m/z = 715.5 (M+H)+
Step 5. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]im^
yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N,3-dimethylbutanamide (Compound 17)

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert- butoxycarbonyl)(methyl)amino)cyclobutyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro- 1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (17c) and (S)-2- ((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 7.89-7.84 (m, 1 H), 7.79- 7.76 (m, 4H), 7.68-7.64 (m, 2H), 7.62-7.60 (m, 3H), 4.36-4.34 (m, 1 H), 4.26-4.24 (m, 2H), 4.04-4.01 (m, 1 H), 3.94-3.95 (m, 1 H), 3.67 (s, 3H), 3.65 (s, 3H), 3.51 -3.49 (m, 1 H), 3.40 (s, 3H), 3.20-3.14 (m, 3H), 2.93-2.91 (m, 2H), 2.69-2.65 (m, 2H), 2.48-2.41 (m, 1 H), 2.37-2.31 (m, 2H), 2.19-1 .95 (m, 5H), 1 .04-0.89 (m, 12H); ); LCMS: m/z = 829.5 (M+H)+.
Example 20: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N,3- dimethylbutanamide (Compound 18):
Step 1 : (S)-tert-butyl 2-(7-(4-(2-(1 -((tert-butoxycarbonyl)(methyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (18a).

Synthesized from tert-butyl methyl(1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (17b) and (S)-tert-butyl 2-(7-
bromo-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 3, Example 3. LCMS: m/z = 713.4 (M+H)+
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)-N,3-dimethylbutanamide (Compound 18)

Synthesized from (S)-tert-butyl 2-(7-(4-(2-(1 -((tert- butoxycarbonyl)(methyl)amino)cyclobutyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-1 H- naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (18a) and (S)-2-
((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3.
1 H-NMR (400 MHz, CD3OD): δ 8.64-8.43 (m, 1 H), 8.34-8.26 (m, 1 H), 7.96-7.93 (m, 1 H), 7.89-7.87 (m, 2H), 7.81 -7.76 (m, 3H), 7.69-7.61 (m, 3H), 4.89-4.30 (m, 2H), 4.29-4.02 (m, 2H), 4.00-3.78 (m, 3H), 3.70-3.68 (m, 1 H), 3.67 (s, 3H), 3.65 (s, 3H), 3.40 (s, 3H), 2.96- 2.94 (m, 2H), 2.68-2.66 (m, 2H), 2.51 -2.48 (m, 1 H), 2.37-2.35 (m, 2H), 2.17-1 .92 (m, 5H), 1 .08-0.89 (m, 12H); LCMS: m/z = 827.3 (M+H)+.
Example 21 : Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclopentyl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide (Compound 19):
Step 1 : 8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert-butoxycarbonyl) amino)cyclopentanecarboxylate (19a)

Step 2: tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)cyclopentyl)carbamate (19b)

8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert-butoxycarbonyl)amino) cyclopentanecarboxylate (19a) (0.41 g, 0.875 mmol) was dissolved in 10 mL of toluene and ammonium acetate (1 .35 g, 17.51 mmol) was added and the reaction mixture was heated at 1 10 °C for 12 h. The reaction mass was cooled to room temperature and water was added and the organics were extracted with 50 mL ethyl acetate twice. The combined ethyl acetate extracts were dried over Na2S04 and concentrated in the rotavap to obtain a brown solid mass which was purified by column chromatography (silica gel; 40-50% EtOAc/hexane) to yield a pale brown colored solid (0.12 g, 31 %). 1 H-NMR (400 MHz, CDCI3): δ 10.69 (brs, 1 H), 7.23-7.21 (m, 3H), 5.16 (brs, 1 H) 4.31 -4.28 (m, 2H), 3.14 (s, 2H), 2.55-2.45 (m, 2H), 2.32-2.20 (m, 2H), 1 .85-1 .82 (m, 4H), 1 .46 (s, 9H); LCMS: m/z = 448.3 (79Br) & 450.3 (81 Br) (M+H)+.
Step 3: Compoun

Synthesized from tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-
2- yl)cyclopentyl)carbamate (19b) and tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6b) by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.73-7.38 (m, 10H), 5.05-4.98 (m, 2H), 4.38-4.28 (m, 2H), 3.50-3.48 (m, 2H), 2.58-2.21 (m, 8H), 1 .96-1 .85 (m, 8H), 1 .49-1 .45 (m, 18H); LCMS: m/z = 745.4 (M+H)+
Step 4: (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)-
3- methylbutanamido)cyclopentyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepi -d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 19)

Synthesized from compound 19c and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 8.72-7.75 (m, 4H), 7.58-7.54 (m, 2H), 7.43-7.41 (m, 2H), 7.37-7.34 (m, 2H), 4.35-4.27 (m, 4H), 3.93-3.85 (m, 2H), 3.71 (s, 3H), 3.75 (s, 3H), 3.20-3.10 (m, 2H), 2.67-1 .92 (m, 16H), 1 .32-1 .30 (m, 2H), 1 .02-0.95 (m, 12H); LCMS: m/z = 859.4 (M+H)+.
Example 22: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-(1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclohexyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopentyl)-3-methylbutanamide (Compound 20):
Step 1 : 8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert-butoxycarbonyl) amino)cyclohexanecarboxylate (20a)

Synthesized from 4,8-dibromo-3,4-dihydrobenzo[b]oxepin-5(2H)-one and 1 -((tert- butoxycarbonyl)amino)cyclohexanecarboxylic acid by following an analogous procedure described in Step 1 , Example 21 . LCMS: m/z = 482.3 (79Br) & 484.3 (81 Br) (M+H)+.
Step 2: tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)cyclohexyl)carbamate (20b)

Synthesized from 8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert- butoxycarbonyl)amino)cyclohexanecarboxylate (20a) by following an analogous procedure described in Step 2, Example 21 . 1 H-NMR (400 MHz, CDCI3): δ 10.60 (brs, 1 H), 7.23-7.18 (m, 3H), 4.80 (brs, 1 H), 4.31 -4.29 (m, 2H), 3.20-3.10 (m, 2H), 2.40-2.20 (m, 2H), 2.15-2.05 (m, 2H), 1 .72-1 .65 (m, 6H), 1 .46 (s, 9H); LCMS: m/z = 462.3 (79Br) & 464.3 (81 Br) (M+H)+.
Step 3: Compoun

Synthesized from tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol- 2-yl)cyclohexyl)carbamate (20b) and tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)carbamate (6b) by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz,
CDCI3): δ 7.75-7.43 (m, 10H), 5.43-5.41 (m, 1 H), 4.38-4.36 (m, 2H), 3.27-3.25 (m, 2H), 2.59-1 .89 (m, 16H), 1 .71 -1 .66 (m, 2H), 1 .48-1 .25 (m, 18H); LCMS: m/z = 759.4 (M+H)+
Step 4: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanamido)cyclohexyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol^ yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 20)

Synthesized from compound (20c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.73-7.61 (m, 4H), 7.56-7.54 (m, 2H), 7.44-7.41 (m, 2H), 7.37-7.35 (m, 2H), 4.35-4.30 (m, 2H), 3.96-3.92 (m, 2H), 3.75 (s, 3H), 3.70 (s, 3H), 3.17-3.14 (m, 4H), 2.57-2.55 (m, 4H), 2.33-1 .30 (m, 16H), 1 .06-1 .01 (m, 12H); LCMS: m/z = 873.5 (M+H)+.
Example 23: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclohexyl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 21 ):
Step 1 : Compoun

Synthesized from tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol- 2-yl)cyclohexyl)carbamate (20b) and tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12b) by
following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): 6 7.73-7.38 (m, 10H), 4.89-4.85 (m, 2H), 4.39-4.36 (m, 2H), 3.25-3.18 (m, 2H), 2.55-2.1 1 (m, 10H), 1 .73-1 .70 (m, 10H), 1 .48-1 .41 (m, 18H); LCMS: m/z = 773.4 (M+H)+
Step 2. (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanamido)cyclohexyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 21 )

Synthesized from compound (21 a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 7.95-7.33 (m, 10H), 4.36-4.28 (m, 2H), 4.02-3.94 (m, 2H), 3.74 (s, 3H), 3.70 (s, 3H), 3.19-3.12 (m, 2H), 2.59-2.57 (m, 4H), 2.29-2.20 (m, 4H), 1 .71 -1 .61 (m, 16H), 1 .07-1 .01 (m, 12H); LCMS: m/z = 887.5 (M+H)+.
Example 24: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclohexyl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide (Compound 22):
Step 1 : Compoun

Synthesized from tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol- 2-yl)cyclopentyl)carbamate (19b) and tert-butyl (1 -(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12b) by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz,
CDCI3): δ 7.72-7.47 (m, 10H), 4.99-4.96 (m, 2H), 4.39-4.36 (m, 2H), 3.43-3.41 (m, 2H), 2.52-2.46 (m, 6H), 2.25-1 .52 (m, 12H), 1 .49-1 .25 (m, 18H); LCMS: m/z = 759.4 (M+H)+
Step 2. (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2-((methoxycarbonyl)amino)- 3-methylbutanamido)cyclohexyl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepi -d]imidazol-2-yl)cyclopentyl)-3-methylbutanarriide (Compound 22)

Synthesized from compound (22a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 8.08-8.06 (m, 1 H), 7.82-7.80 (m, 1 H), 7.74-7.72 (m, 4H), 7.62-7.60 (m, 2H), 7.58-7.54 (m, 2H), 7.45-7.42 (m, 1 H), 7.35-7.34 (m, 1 H), 4.62-4.55 (m, 2H), 4.41 -4.32 (m, 2H), 4.02-4.00 (m, 2H), 3.85-3.83 (m, 2H), 3.74 (s, 3H), 3.70 (s, 3H), 3.18-3.14 (m, 4H), 2.67-1 .60 (m, 14H), 1 .52-1 .48 (m, 2H), 1 .03-0.99 (m, 12H); LCMS: m/z = 873.5 (M+H)+.
Example 25: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)thiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide (Compound 23):
Step 1 : tert-butyl (1 -(5-(5-bromothiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl) carbamate (23a)
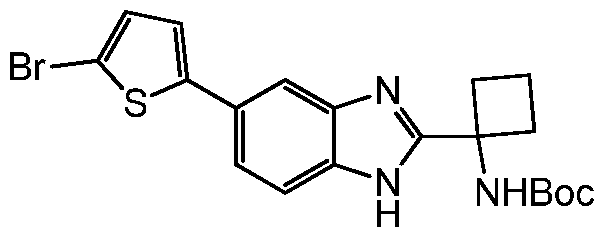
Synthesized from tert-butyl (1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)carbamate (1 b) and 2,5-dibromothiophene by following an
analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 10.60 (brs, 1 H), 7.94-7.43 (m, 3H), 7.04-7.01 (m, 2H), 5.30 (brs, 1 H), 2.99-2.96 (m, 2H), 2.47- 2.37 (m, 2H), 2.20-1 .96 (m, 2H), 1 .48 (s, 9H); LCMS: m/z = 448.0 (79Br) & 450.0 (81 Br) (M+H)+.
Step 2: (S)-tert-butyl 2-(7-(5-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H-benzo[d] imidazol-6-yl)thiophen- -yl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (23b)

Synthesized from (S)-tert-butyl 2-(7-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (prepared as per example 2) and tert-butyl (1 -(5-(5-bromothiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)carbamate (23a) by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): 6 8.1 1 -7.31 (m, 10H), 5.43-5.40 (m, 1 H), 5.30-5.24 (m, 1 H), 3.58-3.50 (m, 2H), 3.16-3.00 (m, 2H), 2.47-2.43 (m, 2H), 2.32-2.28 (m, 2H), 2.17-1 .74 (m, 4H), 1 .30-1 .20 (m, 18H); LCMS: m/z = 705.3
Step 3: (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-d]imidazol-7-yl)thiophen-2-yl)-1 H- benzo[d]imidazol- -yl)cyclobutyl)-3-methylbutanamide (Compound 23)
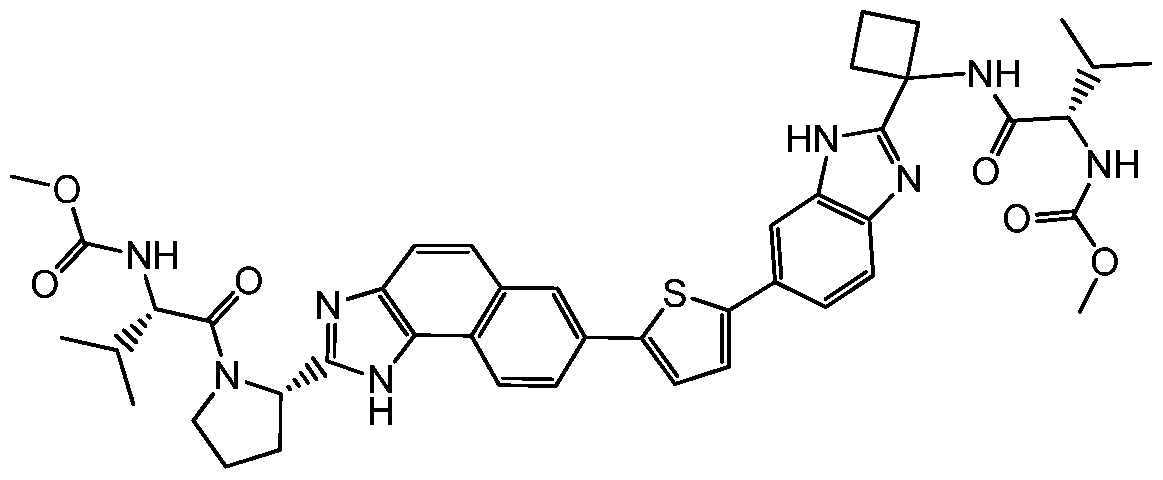
Synthesized from (S)-tert-butyl 2-(7-(5-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- benzo[d]imidazol-6-yl)thiophen-2-yl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (23b) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following
an analogous procedure described in Step 4, Example 3.1H-NMR (400 MHz, CD3OD): δ 8.51 (brs, 1H), 8.30-8.22 (m, 2H), 7.92-7.90 (m, 2H), 7.75-7.70 (m, 2H), 7.62-7.60 (m, 2H), 7.53-7.51 (m, 2H), 7.40-7.39 (m, 1H), 5.39-5.34 (m, 1H), 4.63-4.61 (m, 1H), 4.33-4.31 (m, 1H), 4.26-3.93 (m, 1H), 3.75 (s, 3H), 3.67 (s, 3H), 3.15-2.98 (m, 2H), 2.67-2.45 (m, 4H), 2.31-2.04 (m, 6H), 1.05-0.88 (m, 14H); LCMS: m/z= 819.4 (M+H)+.
Example 26: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1-(6-(5-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1,2-d]imidazol-7-yl)thiophen-2-yl)-1H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 24):
Step 1: tert-butyl (1-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-benzo[d]imidazol- 2-yl)cyclohexyl)carbamate (24a

Synthesized from tert-butyl (1-(5-bromo-1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12a) by following an analogous procedure described in Step 2, Example 1.1H-NMR (400 MHz, CDCI3): δ 7.69 (d, J = 8Hz, 1H), 7.56-7.45 (m, 2H), 4.89 (brs, 1H), 2.44-2.41 (m, 2H), 2.26-2.22 (m, 2H), 1.74-1.65 (m, 4H), 1.63-1.58 (m, 2H), 1.45 (s, 9H), 1.27-1.25 (m, 12H); LCMS: m/z=441.9 (M+H)+
Step 2. tert-butyl (1-(5-(5-bromothiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl) carbamate (24b)

Synthesized from tert-butyl (1-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- benzo[d]imidazol-2-yl)cyclohexyl)carbamate (24a) and 2,5-dibromothiophene by following an analogous procedure described in Step 3, Example 3. 1H-NMR (400 MHz, CDCI3): δ 10.91 (brs, 1H), 7.74-7.72 (m, 1H), 7.43-7.40 (m, 2H), 7.07-7.03 (m, 2H), 4.88 (brs, 1H),
2.43-2.40 (m, 2H), 2.25-2.21 (m, 2H), 1 .72-1 .65 (m, 4H), 1 .61 -1 .59 (m, 2H), 1 .47 (s, 9H); LCMS: m/z = 476.0 (79Br) & 478.0 (81 Br) (M+H)+.
Step 3: (S)-tert-butyl 2-(7-(5-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)thiophen-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imid
yl)pyrrolidine-1 -carboxylate (24c)

Synthesized from tert-butyl (1 -(5-(5-bromothiophen-2-yl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)carbamate (24b) and (S)-tert-butyl 2-(7-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate [Prepared from Miyaura Boration of (S)-tert-butyl 2-(7-bromo-4,5-dihydro-1 H-naphtho[1 ,2- d]imidazol-2-yl)pyrrolidine-1 -carboxylate] by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): 6 10.90 (brs, 1 H), 7.88-7.48 (m, 8H), 5.30-5.24 (m, 1 H), 3.12-3.03 (m, 2H), 2.87-2.84 (m, 2H), 3.58-3.50 (m, 2H), 3.16-3.00 (m, 2H), 2.47-2.43 (m, 2H), 2.32-2.28 (m, 2H), 2.17-1 .76 (m, 4H), 1 .74-1 .70 (m, 4H), 1 .30-1 .20 (m, 18H); LCMS: m/z = 735.5 (M+H)+.
Step 4. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-7- yl)thiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 24)

Example 27: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1-(6-((2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)ethynyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 25):
Step 1 : (S)-tert-butyl 2-(7-((trimethylsilyl)ethynyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (2

(S)-tert-butyl 2-(7-bromo-4,5-dihydro-3H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (858 mg, 2.051 mmol)) was dissolved in 2 mL degassed THF in a microwave vial and triphenylphosphine (26.9 mg, 0.103 mmol), PdCI2(PPh3)2 (144 mg, 0.205 mmol) and triethylamine (0.43 mL, 3.08 mmol) were added to the solution and stirred for 10 min., after which ethynyltrimethylsilane (0.52 mL, 3.69 mmol) and copper(l) iodide (19.5 mg, 0.103 mmol) were added and heated in an oil bath at 55 °C for 16 h. The reaction mixture was extracted with ethyl acetate and washed with water, and the organic layer was dried with Na2S04 and concentrated. The reddish oil was purified on Combiflash™ using 20% EtOAc-Hexane to obtain the pure compound (800 mg, 90%). 1 H-NMR (400 MHz, CDCI3): δ 10.82 (brs, 1 H), 7.71 -7.62 (m, 1 H), 7.50-7.46 (m, 1 H), 7.36-7.30 (m, 1 H), 5.00-4.94 (brs,
1 H), 3.43-3.41 (m, 2H), 3.01 -2.95 (m, 2H), 2.85-2.82 (m, 2H), 2.19-2.1 1 (m, 2H), 1 .99-1 .95 (m, 2H), 1 .52 (s, 9H), 0.25 (s, 9H); LCMS: m/z = 435.1 (M+H)+.
Step 2: (S)-tert-butyl 2-(7-ethynyl-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine- 1 -carboxylate (25b).

(S)-tert-butyl 2-(7-((trimethylsilyl)ethynyl)-4,5-dihydro-3H-naphtho[1 ,2-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (25a) (800 mg, 1 .836 mmol) was dissolved in 20 mL MeOH and potassium carbonate (761 mg, 5.51 mmol) added and stirred at R.T. for 7 h. TLC showed the formation of a polar spot and then the solvent was evaporated to a minimum volume and the contents were extracted with ethyl acetate and washed with water. The ethyl acetate layer was dried over Na2SC>4 and concentrated to obtain the crude material as a brownish solid, which was purified on Combiflash™ with 30% EtOAc-Hexane to obtain the pure compound (490 mg, 74%). LCMS: m/z = 364.3 (M+H)+.
Step 3: (S)-tert-butyl 2-(7-((2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)ethynyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (25c)

(S)-tert-butyl 2-(7-ethynyl-4,5-dihydro-3H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (25b) (1 10 mg, 0.303 mmol) and tert-butyl (1 -(5-iodo-1 H-benzo[d]imidazol-2- yl)cyclohexyl)carbamate (214 mg, 0.484 mmol) [Synthesized from 1 -((tert- butoxycarbonyl)amino)cyclohexanecarboxylic acid and 4-iodobenzene-1 ,2-diamine according to procedure described in Step 1 , Example 3] were dissolved in 2 mL of degassed THF in a microwave vial and triphenylphosphine (4 mg, 0.015 mmol), PdCI2(PPh3)2 (22 mg, 0.03 mmol), triethylamine (0.1 1 ml, 0.757 mmol) and copper(l) iodide (3 mg, 0.015 mmol) were added to the solution and stirred at 45 °C overnight. The
reaction mixture was extracted with ethyl acetate and washed with water, and the organic layer was dried over Na2S04 and concentrated. The reddish oil was purified on Combiflash™ with 20% EtOAc-Hexane to obtain pure compound (105 mg, 49%). 1 H-NMR (400 MHz, CDCI3): 6 7.73-7.62 (m, 3H), 7.37-7.35 (m, 3H), 4.97-4.87 (m, 2H), 3.51 -3.43 (m, 2H), 3.10-2.95 (m, 4H), 2.68-2.54 (m, 2H), 2.36-2.28 (m, 4H), 2.24-2.16 (m, 2H), 2.10- 2.04 (m, 4H), 1 .78-1 .67 (m, 2H), 1 .56-1 .47 (m, 18H); LCMS: m/z = 677.5 (M+H)+.
Step 5. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-((2-((S)-1 -((S)-2-
((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2- d]imidazol-7-yl)ethynyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide
(Compound 25)

Synthesized from (S)-tert-butyl 2-(7-((2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)ethynyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (25c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD), δ 7.73-7.62 (m, 3H), 7.37-7.35 (m, 3H), 5.16-5.13 (m, 2H), 4.55-4.23 (m, 2H), 4.04-4.01 (m, 2H), 3.95-3.88 (m, 2H), 3.73 (s, 3H), 3.66 (s, 3H), 3.05-2.89 (m, 4H), 2.68-2.54 (m, 2H), 2.38-2.27 (m, 4H), 2.22-2.15 (m, 2H), 2.1 1 -2.04 (m, 4H), 1 .78-1 .67 (m, 2H), 1 .05-0.89 (m, 12H); LCMS: m/z = 791 .5 (M+H)+.
Example 28: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)ethyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 26):
Step 4: (S)-tert-butyl 2-(7-(2-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d] imidazol-6-yl)ethyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (26a)
NHBoc
(S)-tert-butyl 2-(7-((2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H-benzo[d]imidazol-6- yl)ethynyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 -carboxylate (25c) (190 mg, 0.281 mmol) was dissolved in 10 mL MeOH and Pd-C (150 mg, 0.141 mmol) was added and kept under a balloon pressure of hydrogen for 18 hr and aliquots were taken for LCMS analysis. After completion of reaction, the mixture was filtered over a Celite bed and washed with ethyl acetate. The filtrate was concentrated to obtain the product (140 mg, 74%). 1 H-NMR (400 MHz, CDCI3): 6 7.73-7.62 (m, 3H), 7.37-7.35 (m, 3H), 4.97-4.87 (m, 2H), 3.49-3.43 (m, 2H), 3.15-2.95 (m, 4H), 2.94-2.88 (m, 4H), 2.68-2.56 (m, 2H), 2.34-2.28 (m, 4H), 2.24-2.16 (m, 2H), 2.12-2.05 (m, 4H), 1 .75-1 .67 (m, 2H), 1 .56-1.47 (m, 18H); LCMS: m/z = 681 .5 (M+H)+.
Step 5. (S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-7-yl)ethyl)- 1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 26

Synthesized from (S)-tert-butyl 2-(7-(2-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)ethyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (26a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1 H-NMR (400 MHz, CD3OD): δ 7.55-7.45 (m, 2H), 7.07-7.05 (m, 2H), 7.01 -6.98 (m, 2H), 5.14-5.14 (m, 2H), 4.55-4.43 (m, 2H), 4.04-4.01 (m, 2H), 3.95-3.88 (m, 2H), 3.73 (s, 3H), 3.66 (s, 3H), 3.05-2.89 (m, 4H), 2.80-2.72 (m, 4H), 2.68-2.54 (m, 2H), 2.38-2.27 (m, 4H), 2.22-2.15 (m, 2H), 2.1 1 -2.04 (m, 4H), 1 .78-1 .67 (m, 2H), 1 .01 -0.88 (m, 12H); LCMS: m/z = 796.6 (M+H)+.
Example 29: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(5-(5-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)thiazol-2-yl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide (Compound 27):
Synthesis of (SHert-butyl 2-(8-(4.4.5.5-tetramethyl-1 .3.2-dioxaborolan-2-vh-4.5-dihvdro- 1 H-benzo[2,31oxepino[4,5-d1imidazol-2-yl)pyrrolidine-1 -carboxylate 27a' :

15 mL of dioxane was degassed by passing nitrogen gas for 10 min. in a microwave vial, and then 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1 ,3,2-dioxaborolane) (468 mg, 1 .84 mmol), (S)-tert-butyl 2-(8-bromo-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine- 1 -carboxylate (400 mg, 0.92 mmol) (ref. WO2009/102633 A1 ), potassium acetate (253 mg, 2.58 mmol), PdCI2(dppf)-CH2Cl2 adduct (75 mg, 0.10 mmol) and tricyclohexylphosphine (27 mg, 0.10 mmol) were added. The vial was sealed and irradiated under microwave at 1 15 °C for 45 min. After cooling, the reaction mixture was added to water and the reaction mass was extracted with 20 % CHCI3-MeOH and dried over Na2S04 and concentrated. Purification was done using Combiflash™ by eluting with 2-5 % MeOH in DCM to yield the title compound as a white foam (310 mg, 68%). 1 H-NMR (400 MHz, CDCI3): 6 8.24 (brs, 1 H), 7.52-7.46 (m, 3H), 5.05-4.97 (brs, 1 H), 4.28-4.26 (m, 2H), 3.43-3.41 (m, 2H), 3.23- 3.20 (m, 2H), 2.18-2.15 (m, 2H), 1 .68-1 .54 (m, 2H), 1 .51 (s, 9H), 1 .33-1 .28 (m, 12H); LCMS: m/z = 482.3 (M+H)+
Step 1 : tert-butyl (1 -(5-(thiazol-2- -1 H-benzo[d]imidazol-2-yl)cyclohexyl)carbamate (27a)

Synthesized from tert-butyl (1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- benzo[d]imidazol-2-yl)cyclohexyl)carbamate (12b) and 2-bromothiazole by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 1 1 .10
(brs, 1 H), 7.85 (d, J = 3.2Hz, 1 H), 7.71 -7.66 (m, 1 H), 7.57-7.54 (m, 1 H), 7.50-7.45 (m, 1 H), 7.30 (d, J = 3.2Hz, 1 H), 4.91 (brs. 1 H), 2.40-2.20 (m, 2H), 2.15-2.05 (m, 2H), 1 .72-1 .65 (m, 6H), 1 .47-1 .42 (m, 9H); LCMS: m/z = 399.2 (M+H)+. Step 2. tert-butyl (1 -(5-(5-bromothiazol-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl) carbamate (27b)


Synthesized from tert-butyl (1 -(5-(5-bromothiazol-2-yl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)carbamate (27b) and (S)-tert-butyl 2-(8-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)pyrrolidine-1 - carboxylate (27a') by following an analogous procedure described in Step 3, Example 3. 1 H-NMR (400 MHz, CDCI3): δ 1 1 .20 (brs, 2H), 7.98 (s, 1 H), 7.91 -7.87 (m, 3H), 7.44-7.39
(m, 3H), 5.01 -4.98 (m, 1 H), 4.34-4.32 (m, 2H), 3.52-3.48 (m, 2H), 3.25-3.23 (m, 2H), 2.99- 2.87 (m, 2H), 2.54-2.41 (m, 2H), 2.23-2.19 (m, 2H), 2.05-1 .97 (m, 2H), 1 .77-1 .68 (m, 2H), 1 .53-1 .47 (m, 18H), 1 .29-1 .25 (m, 4H); LCMS: m/z = 752.4 (M+H)+.
Step 4. (S)-2-(methoxycarbonyl)amino-N-(1 -(5-(5-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imid 8-yl)thiazol-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 27)

Synthesized from (S)-tert-butyl 2-(8-(2-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- benzo[d]imidazol-5-yl)thiazol-5-yl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)pyrrolidine-1 -carboxylate (27c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 3. 1H-NMR (400 MHz, CD3OD): 6 8.05 (s, 1 H), 7.88-7.80 (m, 2H), 7.69-7.56 (m, 2H), 7.44-7.34 (m, 2H), 5.15-5.1 1 (m, 1 H), 4.45-4.28 (m, 4H), 4.02-3.99 (m, 2H), 3.75 (s, 3H), 3.66 (s, 3H), 3.54- 3.49 (m, 1 H), 3.15-3.12 (m, 2H), 2.78-2.63 (m, 2H), 2.38-2.02 (m, 10H), 1 .72-1 .61 (m, 4H), 1 .03-0.84 (m, 12H); LCMS: m/z = 866.4 (M+H)+.
Example 30: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide (Compound 28):
Step 1 : (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-4,5-dihydro- 1 H-benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (28a)

tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol-2- yl)cyclopentyl)carbamate (19b) (154 mg, 0.35 mmol) and (S)-tert-butyl 2-(5-(4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (140 mg, 0.28 mmol) [Prepared according to procedure reported in WO 2010132601 A1 ] was added to a degassed mixture of DME-H20 (2:1 , 3 mL) in a microwave vial followed by Na2C03 (121 mg, 1 .14 mmol) and a catalytic amount of Pd(PPh3)4 (7 mol%). The mixture was subjected to microwave at 120 °C for 30 min. and the reaction mass was extracted with ethyl acetate and washed with water. The ethyl acetate extracts were dried over Na2S04 and concentrated in the rotavap to obtain the crude product which was purified by column chromatography (silica gel; 2-3% MeOH/DCM) to yield a yellowish white solid (60 mg, 28%). 1 H-NMR (400 MHz, CDCI3): δ 10.73 (brs, 1 H), 8.35 (brs, 1 H), 8.04 (brs, 1 H), 7.72-7.67 (m, 4H), 7.55-7.35 (m, 6H), 5.31 (s, 1 H), 5.16-5.12 (m, 1 H), 4.38-4.27 (m, 2H), 3.50-3.40 (m, 2H), 3.19-3.13 (m, 3H), 2.52- 2.49 (m, 3H), 2.32-2.25 (m, 3H), 2.05-1 .90 (m, 1 H), 1 .80-1 .72 (m, 3H), 1 .53-1.42 (m, 18H); LCMS: m/z = 730.9 (M+H)+.
Step 4. (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepin -d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide (Compound 28)

(S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclopentyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (28a) (60 mg, 0.082 mmol) was dissolved in 5 mL of DCM and 2 mL of
trifluoroacetic acid was added to it at room temperature and the solution stirred for 3 h. The volatiles were then removed in the rotavap and the solid obtained was washed with diethyl ether and filtered to obtain the TFA salt of the deprotected compound. The TFA salt in 3 mL of anhydrous DMF at 0 eC, DIPEA (0.105 ml_, 0.603 mmol) was added and the contents were stirred for 10 min., after which (S)-2-((methoxycarbonyl)amino)-3- methylbutanoic acid (33 mg, 0.188 mmol) and HATU (86 mg, 0.226 mmol) were added and the reaction mixture was warmed to room temperature and stirred overnight under nitrogen atmosphere. Crushed ice was added to the reaction mixture and the precipitate formed was filtered, washed with water, n-pentane and dried. Purification using a preparative HPLC yielded a white solid (18 mg, 12%). 1 H-NMR (400 MHz, CD3OD): δ 8.41 (brs, 1 H), 8.08-8.06 (d, J = 9Hz, 1 H), 7.78-7.72 (m, 4H), 7.64-7.55 (m, 3H), 7.44-7.41 (d, J = 9Hz, 1 H), 7.34 (s, 1 H), 5.29-5.25 (m, 1 H), 4.36-4.26 (m, 2H), 4.08-3.91 (m, 2H), 3.85- 3.83 (m, 1 H), 3.70 (s, 3H), 3.67 (s, 3H), 3.16-3.14 (m, 4H), 2.57-2.33 (m, 3H), 2.31 -2.20 (m, 3H), 2.18-2.08 (m, 3H), 2.07-1 .84 (m, 4H), 1 .12-0.81 (m, 12H); LCMS: m/z = 845.5 (M+H)+.
Example 31 : Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 29):
Step 1 : (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (29a)

Synthesized from tert-butyl (1 -(8-bromo-4,5-dihydro-3H-benzo[2,3]oxepino[4,5-d]imidazol- 2-yl)cyclohexyl)carbamate (20b) and (S)-tert-butyl 2-(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 1 , Example 30. 1 H-NMR (400 MHz, CDCI3): δ 10.73 (brs, 1 H), 8.35 (brs, 1 H), 8.04 (brs, 1 H), 7.72-7.67 (m, 4H), 7.55-7.35 (m, 6H), 5.16
(s, 1 H), 4.90-4.80 (m, 1 H), 4.48-4.38 (m, 2H), 3.50-3.44 (m, 2H), 3.26-3.1 1 (m, 4H), 2.37- 2.06 (m, 7H), 1 .80-1 .70 (m, 4H), 1 .53-1 .42 (m, 18H); LCMS: m/z = 744.9 (M+H)+
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepin -d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 29)

Synthesized from (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-4,5- dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)pyrrolidine-1 -carboxylate and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 4, Example 19. 1H-NMR (400 MHz, CD3OD): δ 8.09 (brs, 1 H), 7.78-7.72 (m, 5H), 7.59-7.55 (m, 2H), 7.44-7.41 (m, 2H), 7.33 (brs, 2H), 5.29-5.27 (m, 1 H), 4.42-4.22 (m, 4H), 4.14-4.04 (m, 2H), 3.96-3.94 (m, 2H), 3.70 (s, 3H), 3.67 (s, 3H), 3.16-3.14 (m, 2H), 2.71 -2.67 (m, 1 H), 2.50-2.04 (m, 8H), 1 .80-1 .58 (m, 4H), 1 .12-0.89 (m, 12H); LCMS: m/z = 859.5 (M+H)+.
Example 32: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide (Compound 30):
Step 1 : 6-bromo-1 -oxo-1 ,2,3,4-tetrahydronaphthalen-2-yl 1 -((tert-butoxycarbonyl)amino) cyclobutanecarboxylate (30a)
 Synthesized from 2,6-dibromo-3,4-dihydronaphthalen-1 (2H)-one and 1 -((tert- butoxycarbonyl)amino)cyclobutanecarboxylic acid by following an analogous procedure described in Step 1 , Example 21 . 1 H-NMR (400 MHz, CDCI3): δ 7.89-7.87 (d, J = 8Hz, 1 H); 7.50-7.48 (m, 2H), 5.61 -5.59 (brs, 1 H); 5.32-5.24 (brs, 1 H) 3.26-3.04 (m, 2H), 2.89- 2.74 (m, 2H), 2.41 -2.04 (m, 6H), 1 .48 (s, 9H); LCMS: m/z = 336.1 (79Br) & 338.1 (81Br) (M- Boc)+.
Synthesized from 2,6-dibromo-3,4-dihydronaphthalen-1 (2H)-one and 1 -((tert- butoxycarbonyl)amino)cyclobutanecarboxylic acid by following an analogous procedure described in Step 1 , Example 21 . 1 H-NMR (400 MHz, CDCI3): δ 7.89-7.87 (d, J = 8Hz, 1 H); 7.50-7.48 (m, 2H), 5.61 -5.59 (brs, 1 H); 5.32-5.24 (brs, 1 H) 3.26-3.04 (m, 2H), 2.89- 2.74 (m, 2H), 2.41 -2.04 (m, 6H), 1 .48 (s, 9H); LCMS: m/z = 336.1 (79Br) & 338.1 (81Br) (M- Boc)+.
Step 2: tert-butyl (1 -(7-bromo-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl) carbamate (30b)

Synthesized from 6-bromo-1 -oxo-1 ,2,3,4-tetrahydronaphthalen-2-yl 1 -((tert- butoxycarbonyl)amino)cyclobutanecarboxylate (30a) by following an analogous procedure described in Step 2, Example 21 . LCMS: m/z = 418.1 (79Br) & 420.1 (81 Br) (M+H)+.
Step 3: (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (30c)

Synthesized from tert-butyl (1 -(7-bromo-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2- yl)cyclobutyl)carbamate (30b) and (S)-tert-butyl 2-(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 1 , Example 30. 1 H NMR (400 MHz, CDCI3): δ 10.80-10.60 (brs, 2H), 8.04-7.70 (m, 6H), 7.56-7.50 ( m, 4H), 5.17-5.16 ( m, 1 H), 3.51 -3.41 (m, 2H), 3.93-3.16 (m, 7H), 2.43-2.04 (m, 7H), 1 .54 (s, 9H), 1 .49 (s, 9H); LCMS: m/z 701 .5 (M+H)+
Step 4. (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 30)

Synthesized from (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-4,5- dihydro-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (30c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 2, Example 30. 1 H NMR (400 MHz, CDCI3): δ 7.73-7.71 (m, 6H), 7.60-7.48 (m, 4H), 5.30-5.25 (m, 1 H), 4.28-4.26 (m, 1 H), 4.07-3.84 (m, 3H), 3.67 (s, 3H), 3.36 (s, 3H), 3.13-3.09 (m, 2H), 2.93-2.85 (m, 3H), 2.75-2.22 (m, 6H), 2.19-1 .95 (m, 5H), 0.96-0.94 (m, 6H), 0.90-0.89 (m, 6H); LCMS: m/z = 815.3 (M+H)+.
Example 33: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide
(Compound 31 ):
Step 1 : (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl -1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (31 a)

Synthesized from tert-butyl (1 -(7-bromo-1 H-naphtho[1 ,2-d]imidazol-2- yl)cyclobutyl)carbamate [Prepared from DDQ oxidation of tert-butyl (1 -(7-bromo-4,5- dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)carbamate (30b)] and (S)-tert-butyl 2-(5- (4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-
yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 1 , Example 30. 1 H NMR (400 MHz, CDCI3): δ 1 1 .38 (brs, 1 H), 10.78 (brs, 1 H), 8.73-8.71 (m, 1 H), 8.23-8.20 (m, 1 H), 8.07-7.44 (m, 9H), 6.95-6.93 (m, 1 H), 5.43 (brs, 1 H), 5.18-5.16 (m, 1 H), 3.49-3.45 (m, 2H), 3.12-3.09 (m, 3H), 2.52-2.49 (m, 2H), 2.23-2.06 (m, 5H), 1 .50 (s, 18H); LCMS: m/z = 699.3 (M+H)+
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-1 H- naphtho[1 ,2-d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 31 )

Synthesized from (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (31 a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 2, Example 30. 1 H NMR(400 MHz, CDCI3): δ 8.60 (brs, 1 H), 8.30 (s, 1 H), 7.90-7.85 (m, 4H), 7.81 -7.76 (m, 4H), 7.70-7.50 (m, 2H), 5.31 -5.28 (m, 1 H), 4.28-4.27 (m, 1 H), 4.65 (brs, 1 H), 4.15-3.92 (m, 3H), 3.80 (s, 3H), 3.67 (s, 3H), 3.15-2.57 (m, 5H), 2.49-2.87 (m, 8H), 0.96-0.95 (m, 6H), 0.91 -0.89 (m, 6H); LCMS: m/z = 813.3 (M+H)+.
Example 34: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide (Compound 32):
Step 1 : 6-bromo-1 -oxo-1 ,2,3,4-tetrahydronaphthalen-2-yl 1 -((tert-butoxycarbonyl)amino) cyclohexanecarboxylate (32a)

Synthesized from 2,6-dibromo-3,4-dihydronaphthalen-1(2H)-one and 1-((tert- butoxycarbonyl)amino)cyclohexanecarboxylic acid by following an analogous procedure described in Step 1, Example 21.1H-NMR (400 MHz, CDCI3): δ 7.86-7.79 (m, 1H), 7.53- 7.46 (m, 2H), 5.59-5.55 (m, 1H), 3.19-3.15 (m, 1H), 3.08-3.03 (m, 1H), 2.42-2.38 (m, 1H), 2.28-2.24 (m, 1 H), 2.12-2.04 (m, 2H), 1.99-1.95 (m, 1 H), 1.73-1.65 (m, 2 H), 1.56-1.44 (m, 13H), 1.40-1.27 (m, 2H).
Step 2: tert-butyl (1-(7-bromo-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl) carbamate (32b)

Synthesized from 6-bromo-1-oxo-1 ,2,3,4-tetrahydronaphthalen-2-yl 1-((tert- butoxycarbonyl)amino)cyclohexanecarboxylate (32a) by following an analogous procedure described in Step 2, Example 21.1H-NMR (400 MHz, CDCI3): δ 7.86-7.79 (m, 1H), 7.53- 7.46 (m, 2H), 3.19-3.15 (m, 1H), 3.08-3.03 (m, 1H), 2.42-2.38 (m, 1H), 2.28-2.24 (m, 1H), 2.12-2.04 (m, 2H), 1.99-1.95 (m, 1H), 1.73-1.65 (m, 2 H), 1.56-1.44 (m, 13H), 1.40-1.27 (m, 2H); LCMS: m/z= 445.8 (79Br) & 447.8 (81Br) (M+H)+.
Step 3: (S)-tert-butyl 2-(6-(4-(2-(1-((tert-butoxycarbonyl)amino)cyclohexyl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (32c)

Synthesized from tert-butyl (1-(7-bromo-4,5-dihydro-1 H-naphtho[1,2-d]imidazol-2- yl)cyclohexyl)carbamate and (S)-tert-butyl 2-(5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-
2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 1 , Example 30. LCMS: m/z = 729.5 (M+H)+
Step 4. (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 32)

Synthesized from (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-4,5- dihydro-1 H-naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (32c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 2, Example 30. 1 H-NMR (400 MHz, DMSO-c6): 6 7.99-7.92 (m, 1 H), 7.82-7.69 (m, 4H), 7.59-7.44 (m, 3H), 7.37-7.35 (m, 1 H), 7.24-7.22 (m, 1 H), 5.21 -5.19 (m, 1 H), 4.12-4.05 (m, 2H), 3.93-3.85 (m, 2H), 3.63 (s, 3H), 3.58 (s, 3H), 3.18-3.16 (m, 2H), 3.05-3.03 (m, 2H), 2.85-2.81 (m, 1 H), 2.74-2.67 (m, 2H), 2.26-2.23 (m, 2H), 2.02-1 .86 (m, 5H), 1 .76-1 .74 (m, 1 H), 1 .61 -1 .53 (m, 4H), 1 .31 -1 .23 (m, 2H), 0.95- 0.82 (m, 12H); LCMS: m/z = 843.5 (M+H)+.
Example 35: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6- yl)phenyl)-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide
(Compound 33):
Step 1 : (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (33a)

Synthesized from tert-butyl (1 -(7-bromo-1 H-naphtho[1 ,2-d]imidazol-2- yl)cyclohexyl)carbamate [Prepared from DDQ oxidation of tert-butyl (1 -(7-bromo-4,5- dihydro-1 H-naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)carbamate (32b)] and (S)-tert-butyl 2- (5-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 1 , Example 30. LCMS: m/z = 727.4 (M+H)+
Step 2: (S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-1 H- naphtho[1 ,2-d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide (Compound 33)

Synthesized from (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclohexyl)-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate (33a) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 2, Example 30. 1 H-NMR (400 MHz, CD3OD): δ 8.70-8.68 (d, J=8Hz, 1 H), 8.26-8.24 (m, 1 H), 7.94-7.91 (m, 1 H), 7.89-7.87 (m, 3H), 7.80- 7.75 (m, 3H), 7.72-7.69 (m, 1 H), 7.62-7.57 (m, 2H), 5.21 -5.19 (m, 1 H), 4.12-4.05 (m, 2H), 3.93-3.85 (m, 2H), 3.63 (s, 3H), 3.58 (s, 3H), 3.17-3.15 (m, 2H), 3.05-3.03 (m, 2H), 2.85- 2.81 (m, 1 H), 2.74-2.67 (m, 2H), 2.24-2.21 (m, 2H), 2.02-1 .86 (m, 5H), 1 .76-1 .73 (m, 1 H), 1 .61 -1 .53 (m, 4H), 1 .31 -1 .23 (m, 2H), 0.95-0.82 (m, 12H); LCMS: m/z = 841 .3 (M+H)+.
Example 36: Synthesis of (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1-((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-
yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide (Compound 34):
Step 1 : 8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert-butoxycarbonyl) amino)cyclobutanecarboxylate

Synthesized from 4,8-dibromo-3,4-dihydrobenzo[b]oxepin-5(2H)-one and 1 -((tert- butoxycarbonyl)amino)cyclobutanecarboxylic acid by following an analogous procedure described in Step 1 , Example 21 . 1 H-NMR (400 MHz, CDCI3): δ 7.64-7.62 (m,1 H), 7.30- 7.25 (m, 2H), 5.60-5.55 (m, 1 H), 5.20-5.15 (m, 1 H), 4.63-4.52 (m, 2H), 4.02-3.95 (m, 2H), 2.84-2.61 (m, 4H), 2.18-1 .90 (m, 2H), 1 .44 (s, 9H); LCMS: m/z = 454.3 (79Br) & 456.3 (81 Br) (M+H)+.
Step 2: tert-butyl (1 -(8-bromo-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-2-yl) cyclobutyl)carbamate (34b)

Synthesized from 8-bromo-5-oxo-2,3,4,5-tetrahydrobenzo[b]oxepin-4-yl 1 -((tert- butoxycarbonyl)amino)cyclobutanecarboxylate (34a) by following an analogous procedure described in Step 2, Example 21 . 1 H-NMR (400 MHz, CDCI3): δ 10.50 (brs, 1 H), 7.24-7.20 (m, 3H), 5.16 (brs, 1 H), 4.32-4.30 (m, 2H), 3.20-3.10 (m, 2H), 2.95-2.89 (m, 2H), 2.39-2.34 (m, 2H), 2.15-1 .95 (m, 2H), 1 .46 (s, 9H); LCMS: m/z = 434.3 (79Br) & 436.3 (81 Br) (M+H)+.
Step 3: (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 - carboxylate (34c)

Synthesized from tert-butyl (1 -(8-bromo-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol- 2-yl)cyclobutyl)carbamate (34b) and (S)-tert-butyl 2-(5-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)pyrrolidine-1 -carboxylate by following an analogous procedure described in Step 1 , Example 30. 1 H-NMR (400 MHz, CDCI3): δ 10.73 (brs, 1 H), 8.35 (brs, 1 H), 8.04 (brs, 1 H), 7.72-7.67 (m, 4H), 7.55-7.35 (m, 6H), 5.31 (s, 1 H), 5.18-5.14 (m, 1 H), 4.38-4.27 (m, 2H), 3.50-3.40 (m, 2H), 3.19-3.13 (m, 3H), 2.60- 2.45 (m, 3H), 2.35-2.20 (m, 2H), 2.08-1 .90 (m, 1 H), 1 .84-1 .60 (m, 2H), 1 .53-1.42 (m, 18H); LCMS: m/z = 716.9 (M+H)+
Step 4. (S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2-((methoxycarbonyl) amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H- benzo[2,3]oxepin -d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide (Compound 34)

Synthesized from (S)-tert-butyl 2-(6-(4-(2-(1 -((tert-butoxycarbonyl)amino)cyclobutyl)-4,5- dihydro-1 H-benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)pyrrolidine-1 -carboxylate (34c) and (S)-2-((methoxycarbonyl)amino)-3-methylbutanoic acid by following an analogous procedure described in Step 2, Example 30. 1 H-NMR (400 MHz, CD3OD): 6 7.75-7.70 (m, 2H), 7.60-7.54 (m, 2H), 7.44-7.41 (m, 2H), 7.34-7.30 (m, 2H), 7.22-7.12 (m, 2H), 5.31 -5.29 (m, 1 H), 4.36-4.26 (m, 2H), 4.12-3.91 (m, 2H), 3.85-3.73 (m, 2H), 3.72 (s, 3H), 3.67 (s, 3H), 3.16-3.14 (m, 4H), 2.89-2.55 (m, 3H), 2.38-2.10 (m, 4H), 1 .51 -1 .32 (m, 3H), 1 .32-1 .25 (m, 3H), 1 .12-0.81 (m, 12H); LCMS: m/z = 831 .4 (M+H)+.
Example 37: Biological Activity
Anti-viral activity of the compounds of the invention was monitored using an HCV replicon assay. The Huh7.5/ Con1/SG-Neo(l)hRluc2aUb cell line persistently expressing a bicistronic genotype 1 b replicon in Huh 7.5 cells was obtained from Apath LLC. This cell line was used to test inhibition of replicon levels by test compound using Renilla luciferase enzyme activity readout as a measure of viral replication efficiency.
Briefly, 7000 cells were seeded in 96 well black clear bottom plates and allowed to adhere overnight. The next day each compound was added in triplicate to the cells at the desired concentration with a final DMSO concentration of 0.5%. Cells in media alone and cells incubated without drug with 0.5% DMSO served as controls. The plates were incubated for 72h at 37°C prior to running the luciferase assay. Enzyme activity was measured using Renilla-Glo Luciferase Assay kit from Promega as per the manufacturer's instructions. The following equation was used to generate the percent inhibition value for each test concentration.
Average Control (cells alone +0.5% DMSO) - Average compound valuefcells + drug)
% Inhibition = — 100
Average Control (cells alone+0.5% DMSO)
The EC5o value was determined using GraphPad Prism and the following equation:
Y=Bottom + (Top-Bottom)/(1 +10A((LoglC5o-X)*HillSlope))
EC50 values/% inhibitions of compounds were determined 2-3 times in the replicon assay.
Following table 1 shows EC5o values, for inhibition of genotype 1 b replicon, of the compounds of the invention. Group A compounds exhibited EC50 value between 1 pM to 499 pM, Group B exhibited EC5o value between 500 pM to 999 pM, and Group C exhibited EC50 value of more than 1 nM.
Table 1 :

Claims
1. A compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N- oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrate

RA is independently selected at each occurrence from-

Ring 'A' is selected from 3 to 6 membered carbocycle and 3 to 6 membered heterocycle, the ring 'A' may further be annulated with substituted- or unsubstituted- carbocycle, substituted- or unsubstituted- heterocycle, substituted- or unsubstituted- aromatic carbocycle or substituted- or unsubstituted- aromatic heterocycle; ring D is selected from substituted- or unsubstituted- 5 to 10 membered carbocycles, substituted- or unsubstituted- 5 to 10 membered heterocycles containing 1 to 3 heteroatoms/groups selected from N(R10), S(0)p, O or C(=0), substituted- or unsubstituted- aromatic carbocycle, and 5 to 6 membered substituted- or unsubstituted- aromatic heterocycle containing heteroatoms selected from N, S or O;
Ring Έ' is 5 to 10 membered heterocycle, the ring Έ' may be monocyclic, fused bicyclic, bridged bicyclic or spiro bicyclic;
R1 is selected independently at each occurrence from the group consisting of halogen, substituted- or unsubstituted- Ci_6 alkyl, R10O-, R10aOC(=O)-, and (R 0)(R )NC(=O)-; or two R s taken together may form an oxo (=0) group;
R2 is selected independently at each occurrence from the group consisting of substituted- or unsubstituted- Ci-6 alkyl, R10aC(=O)-, R10aSO2-, R10aOC(=O)-, R10(R11)NC(=O)-, R10aOC(=O)N(R11)C(Ra)(Rb)C(=O)-, R10aOC(=O)N(R11)C(Ra)(Rb)C(Rc)(Rd)C(=O)-,
R10(R11)NC(=O)N(R12)C(Ra)(Rb)C(=O)-,
R10(R11)NC(=O)N(R12)C(Ra)(Rb)C(Rc)(Rd)C(=O)-, R10aSO2N(R11)C(Ra)(Rb)C(=O)-, R 0aSO2N(R11)C(Ra)(Rb)C(Rc)(Rd)C(=O)-, and R10aOC(=O)N(R11)C(Ra)(Rb)SO2-;
R3 is selected from O and N(R13);
R4 is selected independently at each occurrence form CRa(Rb), O, N(R13), C(=0), S(0)p, substituted- or unsubstituted carbocycle, substituted- or unsubstituted- heterocycle, substituted- or unsubstituted- arylene and substituted- or unsubstituted- heteroarylene, wherein, substitutions on carbocycle, heterocycle, arylene, and heteroarylene are selected from the group consisting of halogen, substituted- or unsubstituted- Ci-6 alkyl, and alkyl-O-;
R5 is selected from hydrogen and substituted- or unsubstituted- alkyl;
R6 and R7 are independently selected from the group consisting of hydrogen, halogen, substituted- or unsubstituted- Ci-6 alkyl, and R10O-;
R8 is selected from hydrogen and Ci-6 alkyl;
R9 is selected from the group consisting of hydrogen, Ci-6 alkyl, R10aC(=O)-, R10aSO2-, R10aOC(=O)-, (R10)(R11)NC(=O)-, R10aOC(=O)N(R11)CRa(Rb)C(=O)-, R10aOC(=O)N(R11)CRa(Rb)C(Rc)(Rd)C(=O)-, R10(R11)NC(=O)N(R12)CRa(Rb)C(=O)-, R10(R11)NC(=O)N(R12)CRa(Rb)C(Rc)(Rd)C(=O)-, R10aSO2N(R11)CRa(Rb)C(=O)-, R10aSO2N(R11)CRa(Rb)C(Rc)(Rd)C(=O)-, and R10aOC(=O)N(R11)CRa(Rb)SO2-;
R10, R11 , R11a and R12 are independently selected from hydrogen and substituted- or unsubstituted- Ci-6 alkyl;
R10a is substituted- or unsubstituted- Ci-6 alkyl;
R13 is selected from hydrogen and substituted- or unsubstituted- alkyl group;
Ra, Rb, Rc and Rd, are independently selected from hydrogen, halogen, substituted- or unsubstituted- Ci-6 alkyl, substituted- or unsubstituted- aryl, substituted- or unsubstituted- heteroaryl, substituted- or unsubstituted- cycloalkyi, and substituted- or unsubstituted- heterocyclyl, or Ra, Rb, Rc and Rd together with the carbon atom(s) to which they are attached forming substituted- or unsubstituted- carbocycle, substituted- or unsubstituted- heterocycle; m is an integer ranging between 0 to 2, selected independently at each occurrence; n is an integer ranging between 0 and 2; p is an integer ranging between 0 and 2; when n = 2 and R4 is selected as CRa(Rb) for both the occurrences, two Ras together can form a bond to form a alkenylene linkage or two Ras and two Rbs together can form bonds to form alkynylene linkage;
'alkyl' may be substituted with 1 to 4 substituents selected from the group consisting of oxo, halogen, cyano, aryl, hereroaryl, cycloalkyi, heterocyclyl, (alkyl)C(=0)-, (alkyl)S02-, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, (R14)(H)NC(=0)-, (R14)(alkyl)NC(=0)-, (alkyl)C(=0)N(H)-, R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-;
'cycloalkyi' and 'carbocycle' may be substituted with 1 to 2 substituents selected from the group consisting of oxo, halogen, Ci-6 alkyl, haloalkyl, (alkyl)C(=0)-, (alkyl)S02-, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, R14(H)NC(=0)-, R14(alkyl)NC(=0)-, (alkyl)C(=0)N(H)-, R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-;
'aryl' or 'aromatic carbocycle', may be substituted with 1 to 2 substituents selected from the group consisting of halogen, nitro, cyano, hydroxy, Ci to C6 alkyl, perhaloalkyl, alkyl-O-, perhaloalkyl-O-, alkyl-N(alkyl)-, alkyl-N(H)-, H2N-, alkyl-S02-, alkyl-C(=0)N(alkyl)-, alkyl-C(=0)N(H)-, alkyl-N(alkyl)C(=0)-, alkyl-N(H)C(=0)-, H2NC(=0)-, alkyl-N(alkyl)S02-, alkyl-N(H)S02-, H2NS02-;
'heteroaryl' or 'aromatic heterocycle' may be substituted with 1 to 2 substituents selected from the group consisting of halogen, nitro, cyano, hydroxy, Ci to C6 alkyl, perhaloalkyl, alkyl-O-, perhaloalkyl-O-, alkyl-N(alkyl)-, alkyl-N(H)-, H2N-, alkyl-S02-, alkyl-C(=0)N(alkyl)-, alkyl-C(=0)N(H)-, alkyl-N(alkyl)C(=0)-, alkyl-N(H)C(=0)-, H2NC(=0)-, alkyl-N(alkyl)S02-, and alkyl-N(H)S02-, H2NS02-; ring carbons of 'heterocyclyl' and 'heterocycle' may be substituted with 1 to 2 substituents selected from the group consisting of oxo, halogen, alkyl, R14aO-, (alkyl)OC(=0)-, (alkyl)C(=0)0-, R14(H)NC(=0)-, R14(alkyl)NC(0)-, (alkyl)C(=0)N(H)- , R14(H)N-, R14(alkyl)N-, R14(H)NC(=0)N(H)-, and R14(alkyl)NC(=0)N(H)-; the substituents on ring nitrogen(s) of 'heterocyclyl' and 'heterocycle' are selected from the group consisting of alkyl, (alkyl)C(=0)-, (alkyl)S02-, (alkyl)OC(=0)-, R14(H)NC(=0)-, R14(alkyl)NC(=0)-;
R14 is selected from hydrogen and alkyl;
R14a is selected from hydrogen, alkyl, perhaloalkyl.
2. The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in claim 1 , wherein RA is selected independently at each occurrence from -

and
where, ring A is 3 to 6 membered carbocycle, such that at least once it is selected
as
 The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 and 2, wherein R3 is selected as NH.
The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 and 2, wherein R3 is selected as NH.
The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 -3, wherein R4 is selected from phenylene and five membered heteroarylene containing 1 or 2 heteroatoms selected from N and S, when n is selected as 1 .
The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 -3, wherein R4 is selected as CRaRb, when n is selected as 2 to form an ethylene or ethynylene linkage.
The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 -5, wherein n is selected from 0, 1 and 2.
The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 -6, wherein R5, R6 and R7 are selected as hydrogen.
8. The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 -7, wherein Ring D is selected from the group consisting of aromatic carbocycle, six membered carbocycle, seven membered carbocycle, and seven membered heterocycle containing one heteroatom particularly selected as oxygen.
9. The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 -8, wherein, RA is selected independently at each occurrence from -

where, ring A is 3 to 6 membered carbocycle, such that at least once it is selected
as
 ; R3 is selected as NH; R4 is selected from the group consisting of
; R3 is selected as NH; R4 is selected from the group consisting of
CRaRb, phenylene and five membered heteroarylene containing 1 or 2 heteroatoms selected from N and S; n is selected from 0, 1 and 2; R5, R6 and R7 are selected as hydrogen; and Ring D is selected from the group consisting of aromatic carbocycle, six membered carbocycle, seven membered carbocycle, and seven membered heterocycle containing one heteroatom particularly selected as oxygen.
The compound of formula I, its tautomeric forms, its stereoisomers, its analogues, its prodrugs, its isotopically substituted analogues, its metabolites, its sulfoxides, its N-oxides, its pharmaceutically acceptable salts, its polymorphs, its solvates, its optical isomers, its clathrates or its co-crystals, as claimed in any one of claims 1 -9, wherein the compound is selected from the group consisting of:
x3)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl) 3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclobutyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2 d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopentyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H-
..aphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclopentyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopropyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclopropyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 , 4,5,6- tetrahydrobenzo[3,4]cyclohepta[1 ,2-d]imidazol-8-yl)phenyl)-1 H- benzo[d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide;
x3)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N- ethyl-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)- N,3-dimethylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2- d]imidazol-7-yl)phenyl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-N,3- dimethylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclopentyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5- d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(4-(2-(1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclohexyl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)phenyl)-1 H-benzo[d]imidazol-2- yl)cyclopentyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5- d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-(1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanamido)cyclohexyl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5- d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H-naphtho[1 ,2-
_]imidazol-7-yl)thiophen-2-yl)-1 H-benzo[d]imidazol-2-yl)cyclobutyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(5-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)thiophen-2-yl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-((2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)ethynyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(6-(2-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- naphtho[1 ,2-d]imidazol-7-yl)ethyl)-1 H-benzo[d]imidazol-2-yl)cyclohexyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(5-(5-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-4,5-dihydro-1 H- benzo[2,3]oxepino[4,5-d]imidazol-8-yl)thiazol-2-yl)-1 H-benzo[d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5- d]imidazol-2-yl)cyclopentyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5- d]imidazol-2-yl)cyclohexyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2- yl)cyclobutyl)-3-methylbutanamide;
x3)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]irriidazol-2-yl)cyclobutyl)-3- methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-naphtho[1 ,2-d]imidazol-2- yl)cyclohexyl)-3-methylbutanamide;
(S)-2-(methoxycarbonyl)amino-N-(1 -(7-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H- benzo[d]imidazol-6-yl)phenyl)-1 H-naphtho[1 ,2-d]irriidazol-2-yl)cyclohexyl)-3- methylbutanamide; and
(S)-2-(methoxycarbonyl)amino-N-(1 -(8-(4-(2-((S)-1 -((S)-2- ((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1 H- benzo[d]imidazol-6-yl)phenyl)-4,5-dihydro-1 H-benzo[2,3]oxepino[4,5- d]imidazol-2-yl)cyclobutyl)-3-methylbutanamide.
11. A pharmaceutical composition comprising a compound or a combination of compounds according to any one of claims 1 -10 or a pharmaceutically acceptable salt thereof, in combination with a pharmaceutically acceptable carrier or excipient.
12. A method of inhibiting the replication of an RNA-containing virus comprising contacting said virus with a therapeutically effective amount of a compound or combination of compounds of any one of claims 1 -10, or a pharmaceutically acceptable salt thereof.
13. A method of treating or preventing infection caused by an RNA-containing virus comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound or combination of compounds of any one of claims 1 -10, or a pharmaceutically acceptable salt thereof.
14. The method of claim 12, wherein the RNA-containing virus is hepatitis C virus.
15. The method of claim 12, further comprising the step of co-administering one or more agents selected from the group consisting of a host immune modulator and an antiviral agent, or a combination thereof.
16. The method of claim 14, wherein the host immune modulator is selected from the group consisting of interferon-alpha, pegylated-interferon-alpha, interferon-beta, interferon-gamma, consensus interferon, a cytokine, and a vaccine.
17. The method of claim 14, wherein the antiviral agent inhibits replication of HCV by inhibiting host cellular functions associated with viral replication.
18. The method of claim 14, wherein the antiviral agent inhibits the replication of HCV by targeting proteins of the viral genome.
19. The method of claim 14, wherein said antiviral agent is an inhibitor of a HCV viral protein, a replication process or a combination thereof, wherein said targeting protein or replication process is selected from the group consisting of helicase, protease, polymerase, metalloprotease, NS4A, NS4B, NS5A, assembly, entry, and IRES.
20. The method of claim 12, further comprising the step of co-administering an agent or combination of agents that treat or alleviate symptoms of HCV infection selected from cirrhosis and inflammation of the liver.
21. The method of claim 12, further comprising the step of co-administering one or more agents that treat patients for disease caused by hepatitis B (HBV) infection.
22. The method of claim 12, further comprising the step of co-administering one or more agents that treat patients for disease caused by human immunodeficiency virus (HIV) infection.
23. The pharmaceutical composition of claim 1 1 , further comprising an agent selected from interferon, pegylated interferon, ribavirin, amantadine, an HCV protease inhibitor, an HCV polymerase inhibitor, an HCV helicase inhibitor, or an internal ribosome entry site inhibitor.
24. The composition of claim 1 1 , further comprising a cytochrome P450 monooxygenase inhibitor or a pharmaceutically acceptable salt thereof.
25. A method of treating hepatitis C infection in a subject in need thereof comprising coadministering to said subject a cytochrome P450 monooxygenase inhibitor or a pharmaceutically acceptable salt thereof, and a compound of any one of claims 1 - 10 or a pharmaceutically acceptable salt thereof
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG11201400084XA SG11201400084XA (en) | 2011-09-01 | 2012-08-27 | Antiviral compounds |
| IN363MUN2014 IN2014MN00363A (en) | 2011-09-01 | 2012-08-27 | |
| ZA2014/02374A ZA201402374B (en) | 2011-09-01 | 2014-03-31 | Antiviral compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1157/KOL/2011 | 2011-09-01 | ||
| INKO11572011 | 2011-09-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013030750A1 true WO2013030750A1 (en) | 2013-03-07 |
Family
ID=47010666
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2012/054381 WO2013030750A1 (en) | 2011-09-01 | 2012-08-27 | Antiviral compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2013030750A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9326973B2 (en) | 2012-01-13 | 2016-05-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9717712B2 (en) | 2013-07-02 | 2017-08-01 | Bristol-Myers Squibb Company | Combinations comprising tricyclohexadecahexaene derivatives for use in the treatment of hepatitis C virus |
| US9770439B2 (en) | 2013-07-02 | 2017-09-26 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9775831B2 (en) | 2013-07-17 | 2017-10-03 | Bristol-Myers Squibb Company | Combinations comprising biphenyl derivatives for use in the treatment of HCV |
| US10617675B2 (en) | 2015-08-06 | 2020-04-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
Citations (76)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5019369A (en) | 1984-10-22 | 1991-05-28 | Vestar, Inc. | Method of targeting tumors in humans |
| WO2004014852A2 (en) | 2002-08-12 | 2004-02-19 | Bristol-Myers Squibb Company | Iminothiazolidinones as inhibitors of hcv replication |
| WO2006079833A1 (en) | 2005-01-31 | 2006-08-03 | Arrow Therapeutics Limited | Quinazoline derivatives as antiviral agents |
| WO2006133326A1 (en) | 2005-06-06 | 2006-12-14 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| WO2007031791A1 (en) | 2005-09-16 | 2007-03-22 | Arrow Therapeutics Limited | Biphenyl derivatives and their use in treating hepatitis c |
| WO2007070600A2 (en) | 2005-12-12 | 2007-06-21 | Genelabs Technologies, Inc. | N-(5-membered heteroaromatic ring)-amido anti-viral compounds |
| WO2007070556A2 (en) | 2005-12-12 | 2007-06-21 | Genelabs Technologies, Inc. | N-(6-membered aromatic ring)-amido anti-viral compounds |
| WO2007082554A1 (en) | 2006-01-23 | 2007-07-26 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti Spa | Modulators of hcv replication |
| WO2008021936A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021927A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021928A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008048589A2 (en) | 2006-10-13 | 2008-04-24 | Xtl Biopharmaceuticals Ltd | Compounds and methods for treatment of hcv |
| WO2008064218A2 (en) | 2006-11-21 | 2008-05-29 | Smithkline Beecham Corporation | Amido anti-viral compounds |
| WO2008070447A2 (en) | 2006-11-21 | 2008-06-12 | Smithkline Beecham Corporation | Anti-viral compounds |
| WO2008144380A1 (en) | 2007-05-17 | 2008-11-27 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008154601A1 (en) | 2007-06-12 | 2008-12-18 | Genelabs Technologies, Inc. | Anti-viral inhibitors and methods of use |
| WO2009020825A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Process for synthesizing compounds useful for treating hepatitis c |
| WO2009020828A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Crystalline form of methyl ((1s)-1-(((2s)-2-(5-(4'-(2-((2s)-1-((2s)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)-2-pyrrolidinyl)-1h-imidazol-5-yl)-4-biphenylyl)-1h-imidazol-2-yl)-1-pyrrolidinyl)carbonyl)-2-methylpropyl)carbamate dihydrochloride salt |
| WO2009034390A1 (en) | 2007-09-14 | 2009-03-19 | Arrow Therapeutics Limited | Heterocyclic derivatives and their use in treating hepatitis c |
| EP2086995A1 (en) | 2006-11-16 | 2009-08-12 | Brystol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US20090202483A1 (en) | 2008-02-13 | 2009-08-13 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20090202478A1 (en) | 2008-02-13 | 2009-08-13 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| WO2009102318A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009102694A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | Heterocyclic derivatives as hepatitis c virus inhibitors |
| WO2009102325A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Imidazolyl biphenyl imidazoles as hepatitis c virus inhibitors |
| EP2121697A1 (en) | 2007-03-14 | 2009-11-25 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2010002655A2 (en) | 2008-06-25 | 2010-01-07 | Irm Llc | Compounds and compositions as kinase inhibitors |
| WO2010065668A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010132601A1 (en) | 2009-05-13 | 2010-11-18 | Gilead Sciences, Inc. | Antiviral compounds |
| US20110076291A1 (en) | 2009-09-28 | 2011-03-31 | Nicole Blaquiere | Benzoxepin pi3k inhibitor compounds and methods of use |
| US20110195044A1 (en) * | 2009-02-17 | 2011-08-11 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US8008263B2 (en) | 2007-10-10 | 2011-08-30 | Novartis Ag | Organic compounds and their uses |
| US8008264B2 (en) | 2008-04-23 | 2011-08-30 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US8012942B2 (en) | 2009-02-10 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| US8012982B2 (en) | 2004-10-01 | 2011-09-06 | Istituto Di Ricerche Biologia Molecolare P. Angeletti Spa | Modulators of HCV replication |
| US20110217261A1 (en) | 2010-03-04 | 2011-09-08 | Enanta Pharmaceuticals, Inc. | Combination pharmaceutical agents as inhibitors of hcv replication |
| US20110217265A1 (en) | 2008-09-23 | 2011-09-08 | Glenn Jeffrey S | Screening for Inhibitors of HCV Amphipathic Helix (AH) Function |
| WO2011106929A1 (en) | 2010-03-02 | 2011-09-09 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| WO2011106992A1 (en) | 2010-03-02 | 2011-09-09 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| US20110223134A1 (en) | 2010-03-09 | 2011-09-15 | Anilkumar Gopinadhan Nair | Fused tricyclic silyl compounds and methods of use thereof for the treatment of viral diseases |
| US20110237636A1 (en) | 2009-03-30 | 2011-09-29 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110236348A1 (en) | 2007-06-29 | 2011-09-29 | Gilead Sciences, Inc. | Modulators of toll-like receptor 7 |
| US20110250172A1 (en) | 2010-04-09 | 2011-10-13 | Yao-Ling Qiu | Hepatitis c virus inhibitors |
| US20110250176A1 (en) | 2009-10-12 | 2011-10-13 | Bristol-Myers Squibb Company | Combinations of Hepatitis C Virus Inhibitors |
| US20110269956A1 (en) | 2009-11-11 | 2011-11-03 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110274648A1 (en) | 2009-11-11 | 2011-11-10 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110281910A1 (en) | 2009-11-12 | 2011-11-17 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110286961A1 (en) | 2009-12-16 | 2011-11-24 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110294819A1 (en) | 2009-12-30 | 2011-12-01 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| WO2011153396A1 (en) | 2010-06-04 | 2011-12-08 | Enanta Pharmaceuticals, Inc | Hepatitis c virus inhibitors |
| WO2011151652A1 (en) | 2010-06-03 | 2011-12-08 | Arrow Therapeutics Limited | Benzodiazepine compounds useful for the treatment of hepatitis c |
| WO2011151651A1 (en) | 2010-06-03 | 2011-12-08 | Arrow Therapeutics Limited | Benzodiazepine compounds useful for the treatment of hepatitis c |
| WO2011156543A2 (en) | 2010-06-09 | 2011-12-15 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a protein |
| US20120004196A1 (en) | 2009-06-11 | 2012-01-05 | Abbott Labaoratories | Anti-Viral Compounds |
| US8101643B2 (en) | 2009-02-27 | 2012-01-24 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US20120028978A1 (en) | 2009-03-27 | 2012-02-02 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic hcv inhibitors |
| WO2012018325A1 (en) | 2010-08-04 | 2012-02-09 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2012018534A2 (en) | 2010-07-26 | 2012-02-09 | Schering Corporation | Substituted biphenylene compounds and methods of use thereof for the treatment of viral diseases |
| WO2012021591A1 (en) | 2010-08-12 | 2012-02-16 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2012021704A1 (en) | 2010-08-12 | 2012-02-16 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| US20120040962A1 (en) | 2009-03-27 | 2012-02-16 | Presidio Pharmaceuticals, Inc. | Fused ring inhibitors of hepatitis c |
| US20120040977A1 (en) | 2009-02-23 | 2012-02-16 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2012024363A2 (en) | 2010-08-17 | 2012-02-23 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
| US8133884B2 (en) | 2008-05-06 | 2012-03-13 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US8143301B2 (en) | 2009-04-09 | 2012-03-27 | Bristol Myers Squibb Company | Hepatitis C virus inhibitors |
| US8143414B2 (en) | 2009-04-13 | 2012-03-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20120076755A1 (en) | 2010-01-25 | 2012-03-29 | Enanta Pharmaceuticals, Inc. | Hepatitis C Virus Inhibitors |
| WO2012039717A1 (en) | 2010-09-24 | 2012-03-29 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US20120083483A1 (en) | 2009-03-27 | 2012-04-05 | Merck Sharp & Dohme Corp | Inhibitors of hepatitis c virus replication |
| WO2012041227A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds for treating hepatitis c viral infection |
| WO2012041014A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives for treating hepatitis c virus infection |
| US8188132B2 (en) | 2009-02-17 | 2012-05-29 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| US8198449B2 (en) | 2008-09-11 | 2012-06-12 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
-
2012
- 2012-08-27 WO PCT/IB2012/054381 patent/WO2013030750A1/en active Application Filing
Patent Citations (88)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US5019369A (en) | 1984-10-22 | 1991-05-28 | Vestar, Inc. | Method of targeting tumors in humans |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| WO2004014852A2 (en) | 2002-08-12 | 2004-02-19 | Bristol-Myers Squibb Company | Iminothiazolidinones as inhibitors of hcv replication |
| US8012982B2 (en) | 2004-10-01 | 2011-09-06 | Istituto Di Ricerche Biologia Molecolare P. Angeletti Spa | Modulators of HCV replication |
| WO2006079833A1 (en) | 2005-01-31 | 2006-08-03 | Arrow Therapeutics Limited | Quinazoline derivatives as antiviral agents |
| WO2006133326A1 (en) | 2005-06-06 | 2006-12-14 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| US8143288B2 (en) | 2005-06-06 | 2012-03-27 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| WO2007031791A1 (en) | 2005-09-16 | 2007-03-22 | Arrow Therapeutics Limited | Biphenyl derivatives and their use in treating hepatitis c |
| WO2007070600A2 (en) | 2005-12-12 | 2007-06-21 | Genelabs Technologies, Inc. | N-(5-membered heteroaromatic ring)-amido anti-viral compounds |
| WO2007070556A2 (en) | 2005-12-12 | 2007-06-21 | Genelabs Technologies, Inc. | N-(6-membered aromatic ring)-amido anti-viral compounds |
| WO2007082554A1 (en) | 2006-01-23 | 2007-07-26 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti Spa | Modulators of hcv replication |
| WO2008021928A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021927A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021936A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| EP2049116A2 (en) | 2006-08-11 | 2009-04-22 | Brystol-Myers Squibb Company | Hepatitis c virus inhibitors |
| EP2385048A1 (en) | 2006-08-11 | 2011-11-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2008048589A2 (en) | 2006-10-13 | 2008-04-24 | Xtl Biopharmaceuticals Ltd | Compounds and methods for treatment of hcv |
| EP2086995A1 (en) | 2006-11-16 | 2009-08-12 | Brystol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008070447A2 (en) | 2006-11-21 | 2008-06-12 | Smithkline Beecham Corporation | Anti-viral compounds |
| WO2008064218A2 (en) | 2006-11-21 | 2008-05-29 | Smithkline Beecham Corporation | Amido anti-viral compounds |
| EP2121697A1 (en) | 2007-03-14 | 2009-11-25 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| EP2146984A1 (en) | 2007-05-17 | 2010-01-27 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008144380A1 (en) | 2007-05-17 | 2008-11-27 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008154601A1 (en) | 2007-06-12 | 2008-12-18 | Genelabs Technologies, Inc. | Anti-viral inhibitors and methods of use |
| US20110236348A1 (en) | 2007-06-29 | 2011-09-29 | Gilead Sciences, Inc. | Modulators of toll-like receptor 7 |
| WO2009020828A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Crystalline form of methyl ((1s)-1-(((2s)-2-(5-(4'-(2-((2s)-1-((2s)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)-2-pyrrolidinyl)-1h-imidazol-5-yl)-4-biphenylyl)-1h-imidazol-2-yl)-1-pyrrolidinyl)carbonyl)-2-methylpropyl)carbamate dihydrochloride salt |
| WO2009020825A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Process for synthesizing compounds useful for treating hepatitis c |
| WO2009034390A1 (en) | 2007-09-14 | 2009-03-19 | Arrow Therapeutics Limited | Heterocyclic derivatives and their use in treating hepatitis c |
| US8008263B2 (en) | 2007-10-10 | 2011-08-30 | Novartis Ag | Organic compounds and their uses |
| US8093243B2 (en) | 2008-02-12 | 2012-01-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2009102694A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | Heterocyclic derivatives as hepatitis c virus inhibitors |
| WO2009102318A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| EP2250163A1 (en) | 2008-02-12 | 2010-11-17 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US20090202478A1 (en) | 2008-02-13 | 2009-08-13 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20090202483A1 (en) | 2008-02-13 | 2009-08-13 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| WO2009102633A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US8147818B2 (en) | 2008-02-13 | 2012-04-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2009102325A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Imidazolyl biphenyl imidazoles as hepatitis c virus inhibitors |
| US20110293563A1 (en) | 2008-04-23 | 2011-12-01 | Gilead Sciences, Inc. | 1'substituted carba-nucleoside analogs for antiviral treatment |
| US8008264B2 (en) | 2008-04-23 | 2011-08-30 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US8012941B2 (en) | 2008-04-23 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| US8133884B2 (en) | 2008-05-06 | 2012-03-13 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| WO2010002655A2 (en) | 2008-06-25 | 2010-01-07 | Irm Llc | Compounds and compositions as kinase inhibitors |
| US8198449B2 (en) | 2008-09-11 | 2012-06-12 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US20110217265A1 (en) | 2008-09-23 | 2011-09-08 | Glenn Jeffrey S | Screening for Inhibitors of HCV Amphipathic Helix (AH) Function |
| US20110237579A1 (en) | 2008-12-03 | 2011-09-29 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010065668A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| US8012942B2 (en) | 2009-02-10 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| US8188132B2 (en) | 2009-02-17 | 2012-05-29 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| US20110195044A1 (en) * | 2009-02-17 | 2011-08-11 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20120040977A1 (en) | 2009-02-23 | 2012-02-16 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| US8101643B2 (en) | 2009-02-27 | 2012-01-24 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US20120028978A1 (en) | 2009-03-27 | 2012-02-02 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic hcv inhibitors |
| US20120040962A1 (en) | 2009-03-27 | 2012-02-16 | Presidio Pharmaceuticals, Inc. | Fused ring inhibitors of hepatitis c |
| US20120083483A1 (en) | 2009-03-27 | 2012-04-05 | Merck Sharp & Dohme Corp | Inhibitors of hepatitis c virus replication |
| US20110237636A1 (en) | 2009-03-30 | 2011-09-29 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US8143301B2 (en) | 2009-04-09 | 2012-03-27 | Bristol Myers Squibb Company | Hepatitis C virus inhibitors |
| US8143414B2 (en) | 2009-04-13 | 2012-03-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2010132601A1 (en) | 2009-05-13 | 2010-11-18 | Gilead Sciences, Inc. | Antiviral compounds |
| US20120004196A1 (en) | 2009-06-11 | 2012-01-05 | Abbott Labaoratories | Anti-Viral Compounds |
| US20110076291A1 (en) | 2009-09-28 | 2011-03-31 | Nicole Blaquiere | Benzoxepin pi3k inhibitor compounds and methods of use |
| US20110250176A1 (en) | 2009-10-12 | 2011-10-13 | Bristol-Myers Squibb Company | Combinations of Hepatitis C Virus Inhibitors |
| US20110269956A1 (en) | 2009-11-11 | 2011-11-03 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110274648A1 (en) | 2009-11-11 | 2011-11-10 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110281910A1 (en) | 2009-11-12 | 2011-11-17 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110286961A1 (en) | 2009-12-16 | 2011-11-24 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20110294819A1 (en) | 2009-12-30 | 2011-12-01 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20120076755A1 (en) | 2010-01-25 | 2012-03-29 | Enanta Pharmaceuticals, Inc. | Hepatitis C Virus Inhibitors |
| WO2011106992A1 (en) | 2010-03-02 | 2011-09-09 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| WO2011106929A1 (en) | 2010-03-02 | 2011-09-09 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| US20110217261A1 (en) | 2010-03-04 | 2011-09-08 | Enanta Pharmaceuticals, Inc. | Combination pharmaceutical agents as inhibitors of hcv replication |
| US20110223134A1 (en) | 2010-03-09 | 2011-09-15 | Anilkumar Gopinadhan Nair | Fused tricyclic silyl compounds and methods of use thereof for the treatment of viral diseases |
| US20110250172A1 (en) | 2010-04-09 | 2011-10-13 | Yao-Ling Qiu | Hepatitis c virus inhibitors |
| WO2011151652A1 (en) | 2010-06-03 | 2011-12-08 | Arrow Therapeutics Limited | Benzodiazepine compounds useful for the treatment of hepatitis c |
| WO2011151651A1 (en) | 2010-06-03 | 2011-12-08 | Arrow Therapeutics Limited | Benzodiazepine compounds useful for the treatment of hepatitis c |
| US20110300104A1 (en) | 2010-06-04 | 2011-12-08 | Yao-Ling Qiu | Hepatitis c virus inhibitors |
| WO2011153396A1 (en) | 2010-06-04 | 2011-12-08 | Enanta Pharmaceuticals, Inc | Hepatitis c virus inhibitors |
| WO2011156543A2 (en) | 2010-06-09 | 2011-12-15 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a protein |
| WO2012018534A2 (en) | 2010-07-26 | 2012-02-09 | Schering Corporation | Substituted biphenylene compounds and methods of use thereof for the treatment of viral diseases |
| WO2012018325A1 (en) | 2010-08-04 | 2012-02-09 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2012021704A1 (en) | 2010-08-12 | 2012-02-16 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| WO2012021591A1 (en) | 2010-08-12 | 2012-02-16 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2012024363A2 (en) | 2010-08-17 | 2012-02-23 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
| WO2012039717A1 (en) | 2010-09-24 | 2012-03-29 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2012041227A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds for treating hepatitis c viral infection |
| WO2012041014A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives for treating hepatitis c virus infection |
Non-Patent Citations (25)
| Title |
|---|
| "ASHP Handbook on Injectable Drugs, Toissel, 4th ed.,", 1986, pages: 622 - 630 |
| "Greene and Wuts 'protective groups in Organic Synthesis", 1999, WILEY AND SONS |
| "Physicians' Desk Reference, 58th ed.", 2004, THOMSON PDR |
| "Remington's Pharmaceutical Science", 1985, MACK PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences, 18th ed.,", 1990, MACK PUBLISHING COMPANY, pages: 1445 |
| ACC. CHEM. RES., vol. 31, 1998, pages 852 - 860 |
| BANKER AND CHALMERS,: "Pharmaceutics and Pharmacy Practice", 1982, J.B. LIPPINCOTT COMPANY, pages: 238 - 250 |
| BARNES E., WHO FACTSHEET, 2010, Retrieved from the Internet <URL:http://www.who.int/vaccine_research/diseases/virai_cancers/en/index2.htmi> |
| BERGE S.M. ET AL.: "Pharmaceutical Salts, a review article", JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 66, 1977, pages 1 - 19 |
| BIOORG. MED. CHEM. LETT., 2002, pages 1009 - 1011 |
| CANADIAN JOURNAL OF CHEMISTRY, vol. 78, no. 7, 2000, pages 957 - 962 |
| CHEM. REV., vol. 95, 1995, pages 2457 - 83 |
| FRIED MW ET AL., N ENGL J MED., vol. 347, 2002, pages 975 - 982 |
| HOOFNAGLE JH., HEPATOLOGY, vol. 26, 1997, pages 15S - 20 |
| J. AM. CHEM. SOC., vol. 125, no. 22, 2003, pages 6653 - 55 |
| J. ORG. CHEM., vol. 71, no. 1, 2006, pages 379 - 381 |
| JESUDIAN AB; GAMBARIN-GELWAN M; JACOBSON IM., GASTROENTEROLOGY HEPATOL., vol. 8, 2012, pages 91 - 101 |
| JOURNAL OF PHARMACEUTICAL SCIENCE, vol. 66, 1977, pages 2 - 19 |
| MORIISHI K ET AL., ANTIVIR. CHEM. CHEMOTHER., vol. 14, 2003, pages 285 - 297 |
| P.H.EINRICH; STAHLAND CAMILLE; G.WERMUTH: "handbook of pharmaceutical salts properties, selection", 2002, WILEY- VCH |
| PHYSICIANS' DESK REFERENCE, 2004 |
| SZABO E; LOTZ G ET AL., PATHOL. ONCOL. RES., vol. 9, 2003, pages 215 - 221 |
| SZOKA ET AL., ANN. REV. BIOPHYS. BIOENG., vol. 9, 1980, pages 467 |
| THOMSON BJ ET AL., CLIN MICROBIAL INFECT., vol. 11, 2005, pages 86 - 94 |
| WASSERMAN ET AL., CANCER, vol. 36, 1975, pages 1258 - 1268 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9326973B2 (en) | 2012-01-13 | 2016-05-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9717712B2 (en) | 2013-07-02 | 2017-08-01 | Bristol-Myers Squibb Company | Combinations comprising tricyclohexadecahexaene derivatives for use in the treatment of hepatitis C virus |
| US9770439B2 (en) | 2013-07-02 | 2017-09-26 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9775831B2 (en) | 2013-07-17 | 2017-10-03 | Bristol-Myers Squibb Company | Combinations comprising biphenyl derivatives for use in the treatment of HCV |
| US10617675B2 (en) | 2015-08-06 | 2020-04-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2013217229B2 (en) | Antiviral compounds with a heterotricycle moiety | |
| EP2516430B1 (en) | Fused tricyclic compounds and methods of use thereof for the treatment of viral diseases | |
| DK2784075T3 (en) | Hepatitis C viral inhibitors | |
| EP2435424B1 (en) | Antiviral compounds composed of three linked aryl moieties to treat diseases such as hepatitis c | |
| TWI501957B (en) | Hepatitis c virus inhibitors | |
| US8697704B2 (en) | Hepatitis C virus inhibitors | |
| WO2010148006A1 (en) | Hepatitis c virus inhibitors | |
| JP2013518060A (en) | Hepatitis C virus inhibitor | |
| WO2013021337A1 (en) | Antiviral compounds with a fused tricyclic ring | |
| WO2009003009A1 (en) | Substituted pyrrolidine as anti-infectives | |
| CA2753382A1 (en) | Hepatitis c virus inhibitors | |
| EP2575819A1 (en) | Hepatitis c virus inhibitors | |
| JP2014534206A (en) | Hepatitis C virus inhibitor | |
| WO2013052362A1 (en) | Hepatitis c virus inhibitors | |
| MX2013012870A (en) | Hepatitis c virus inhibitors. | |
| CN103880823B (en) | Application as the spiro-compound of hepatitis c inhibitor and its in medicine | |
| WO2013030750A1 (en) | Antiviral compounds | |
| WO2013021344A1 (en) | Imidazole derivatives as antiviral agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12770245 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12770245 Country of ref document: EP Kind code of ref document: A1 |