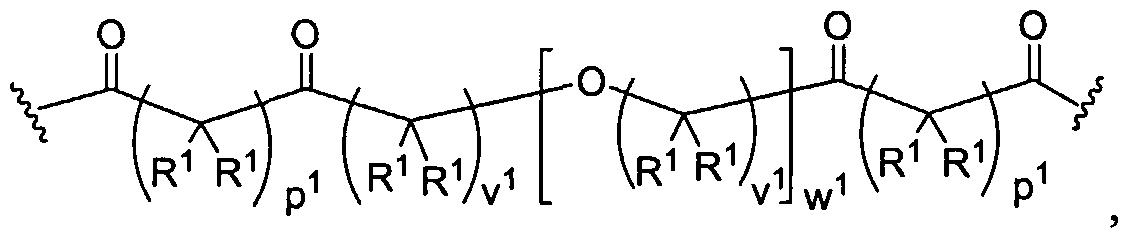
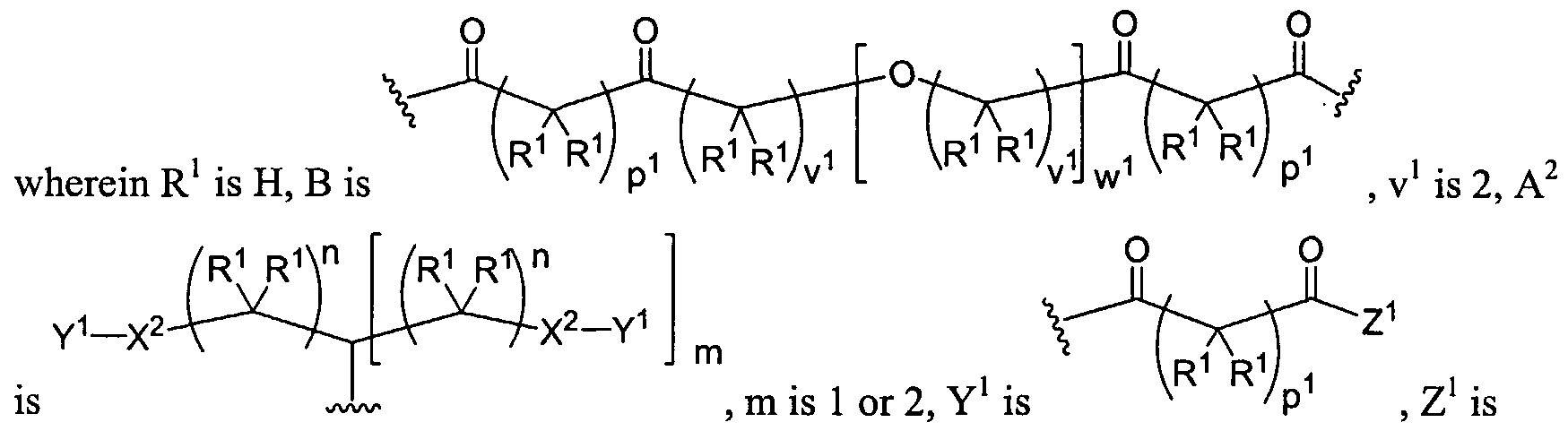
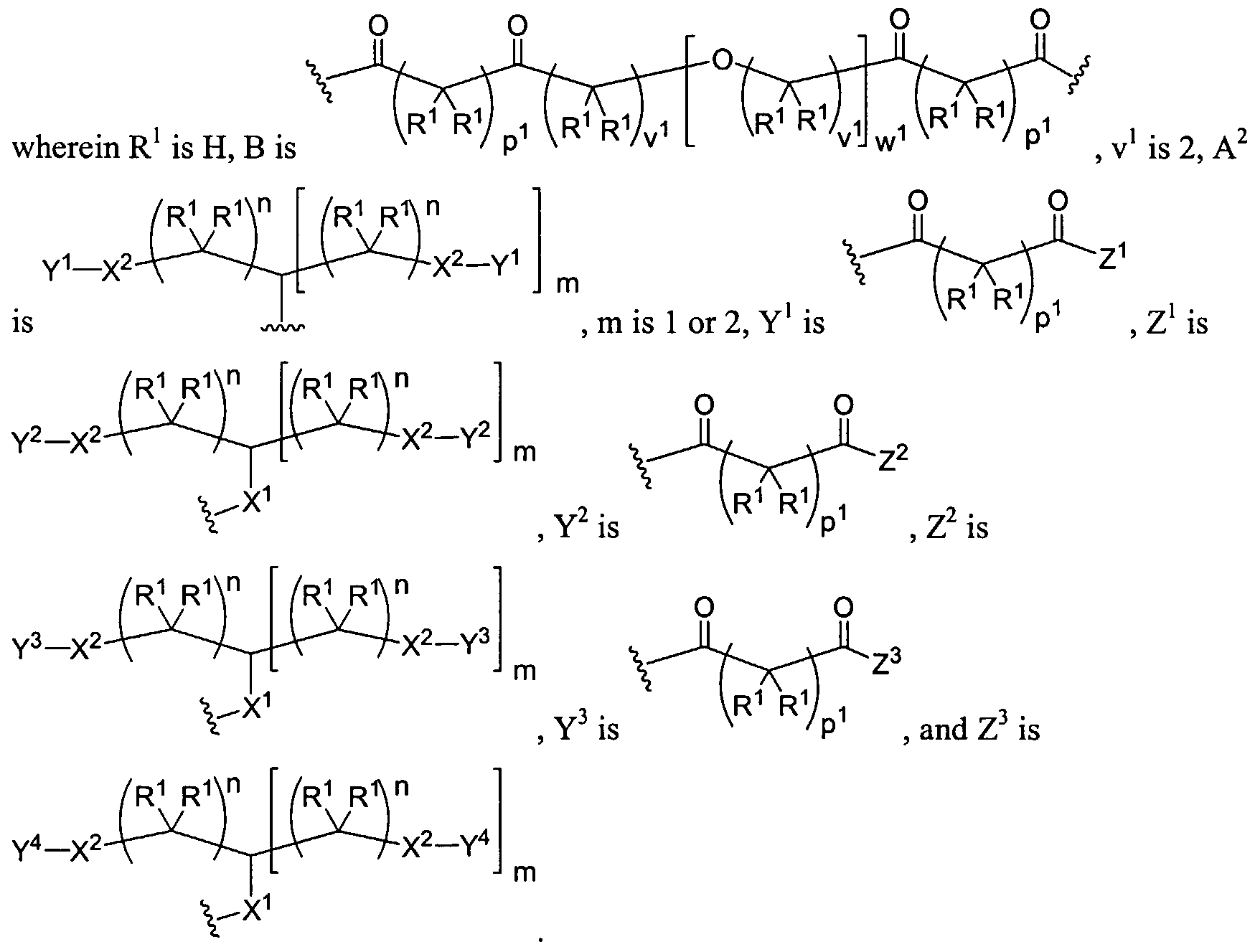
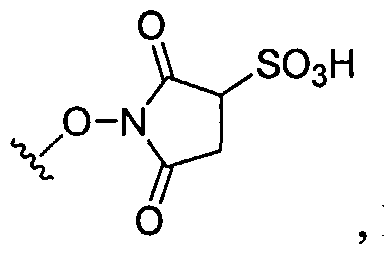
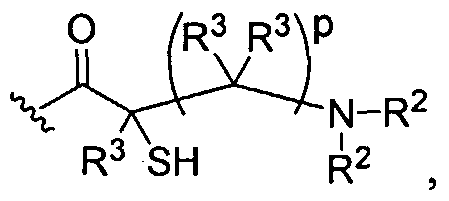
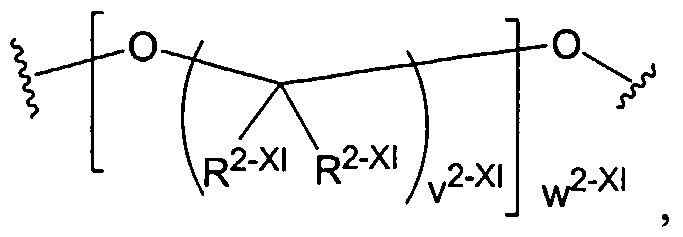
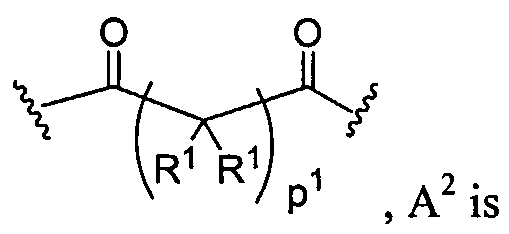
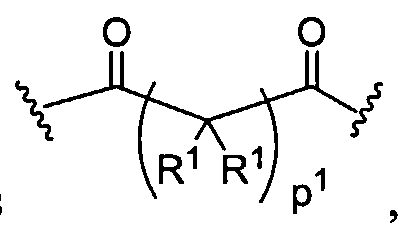
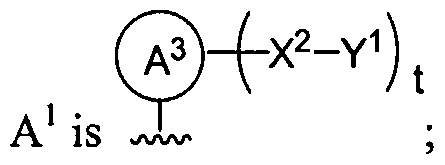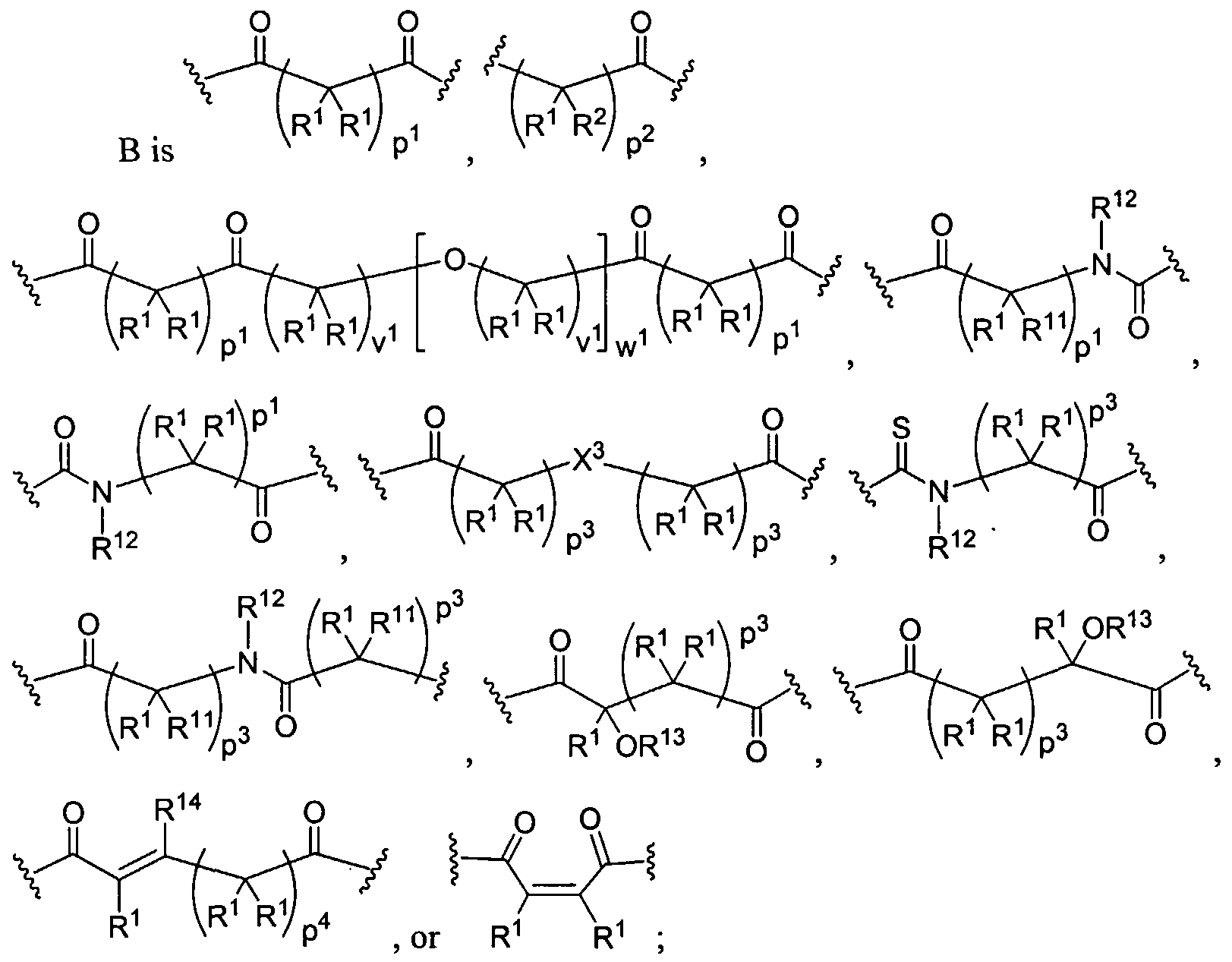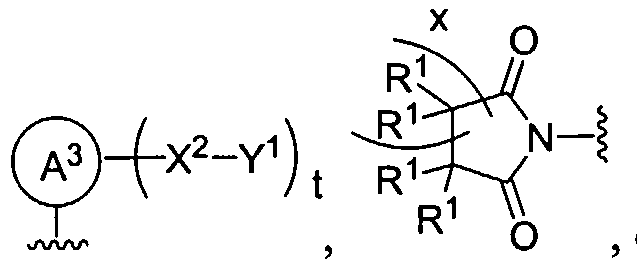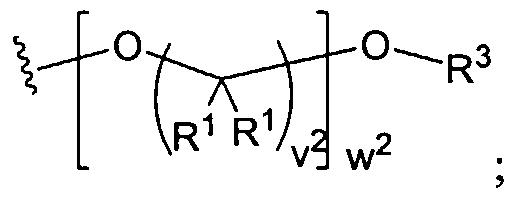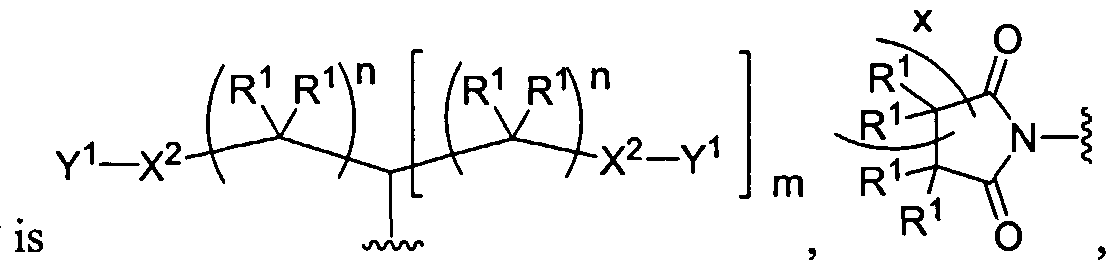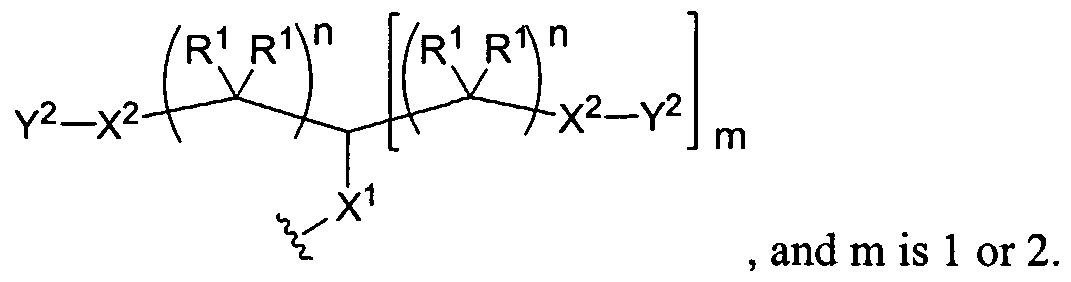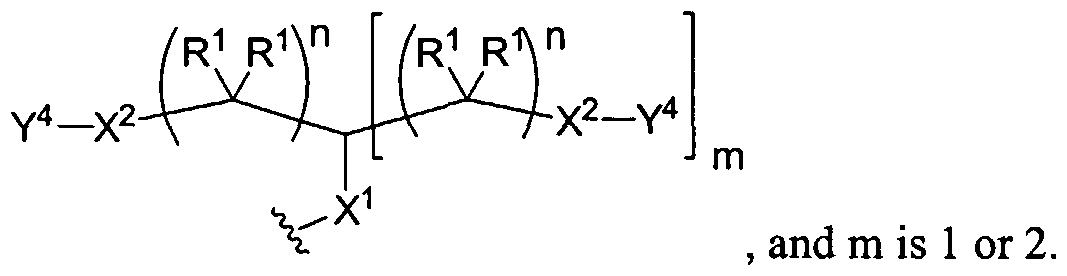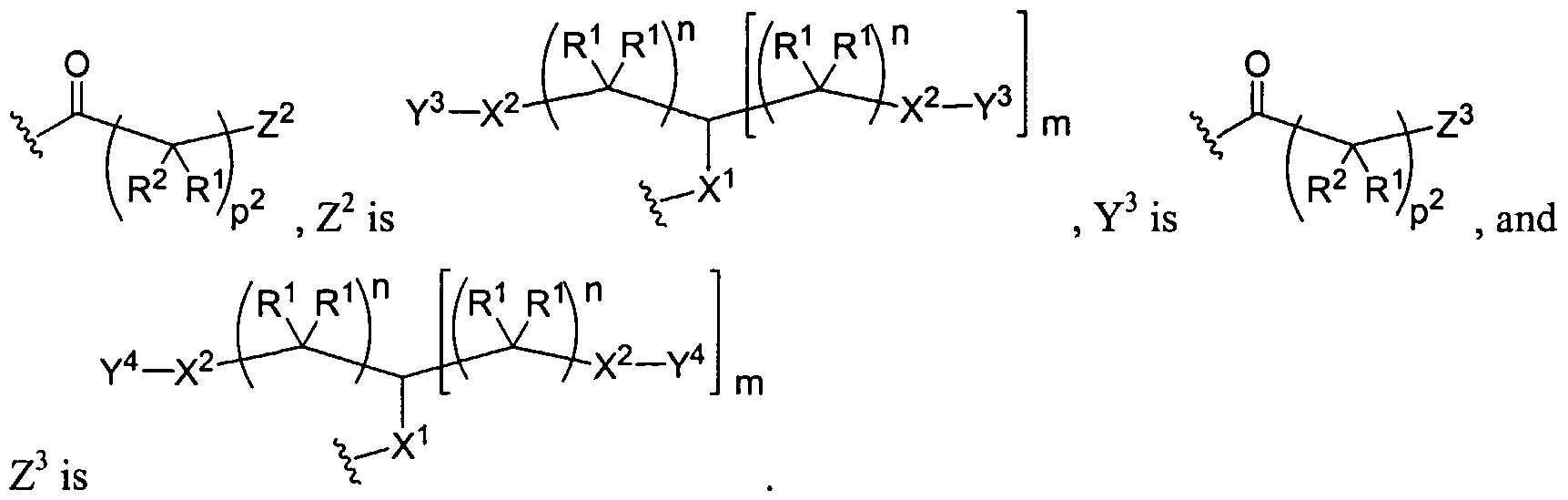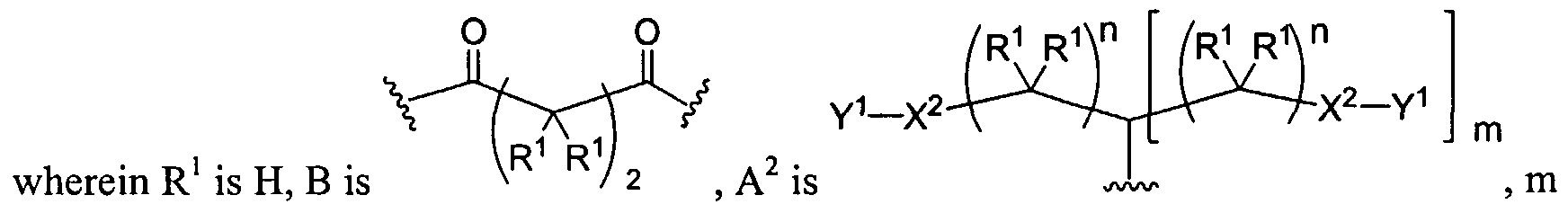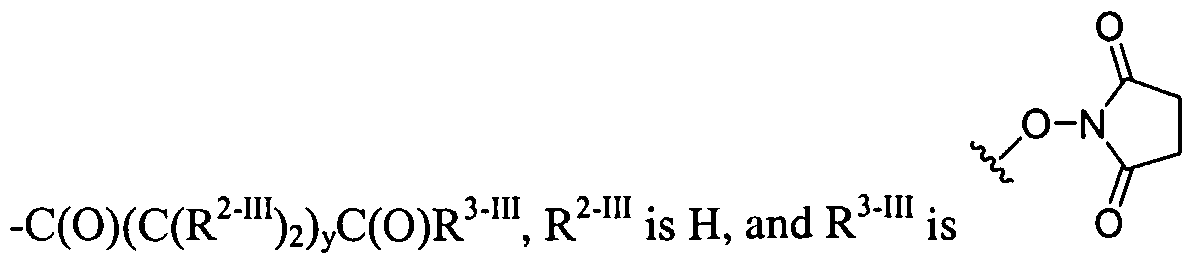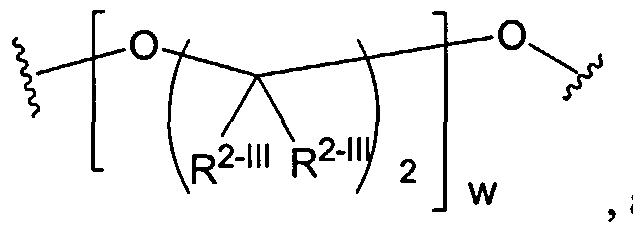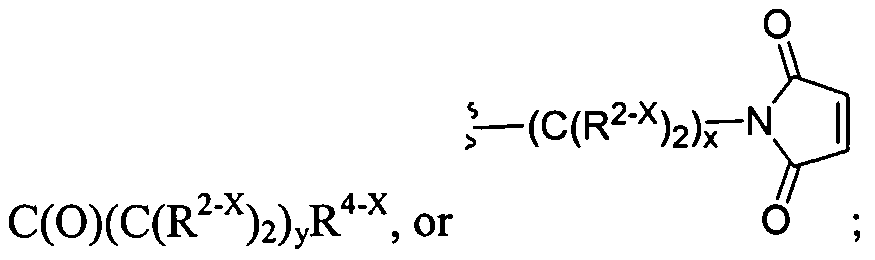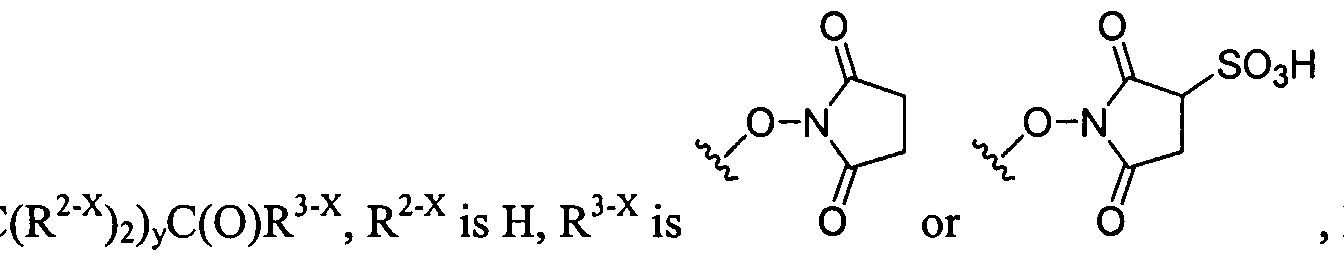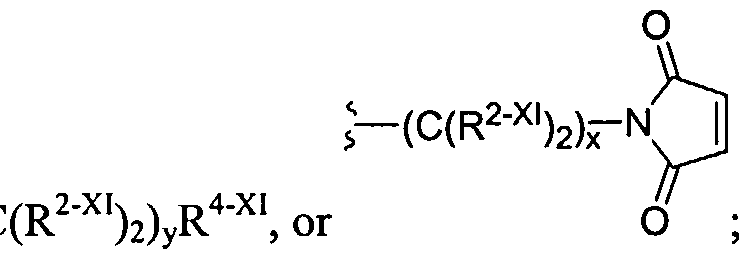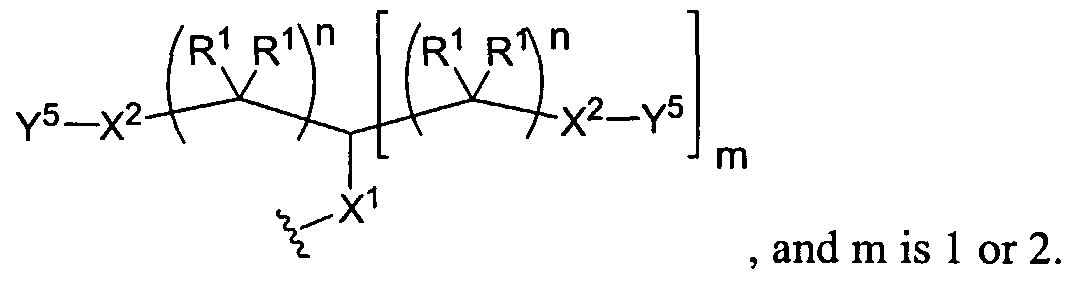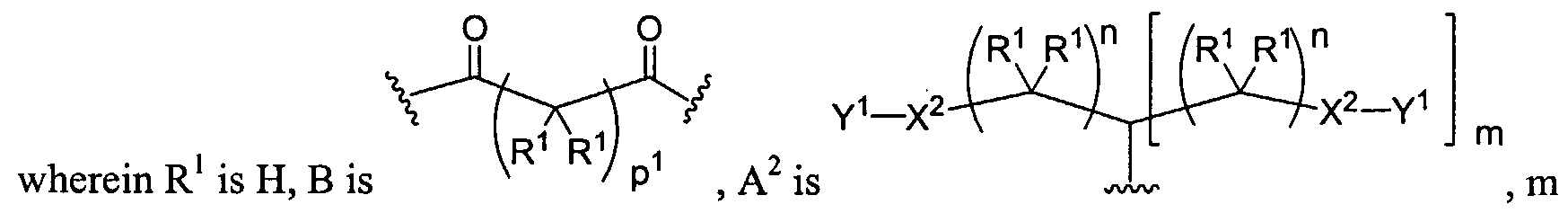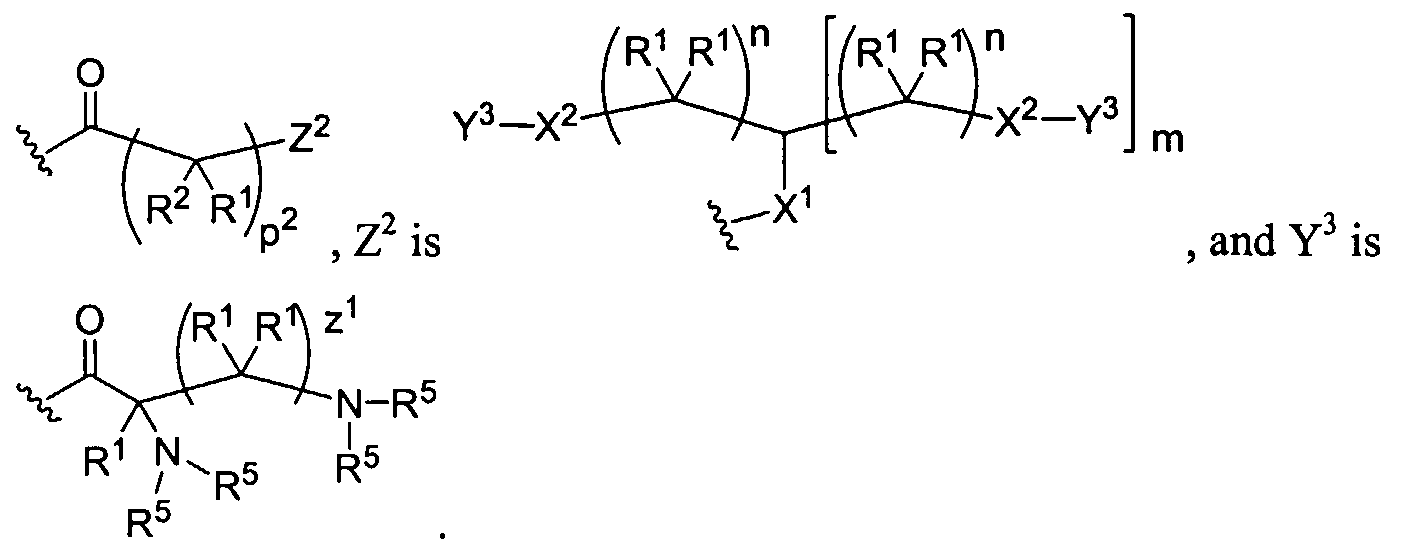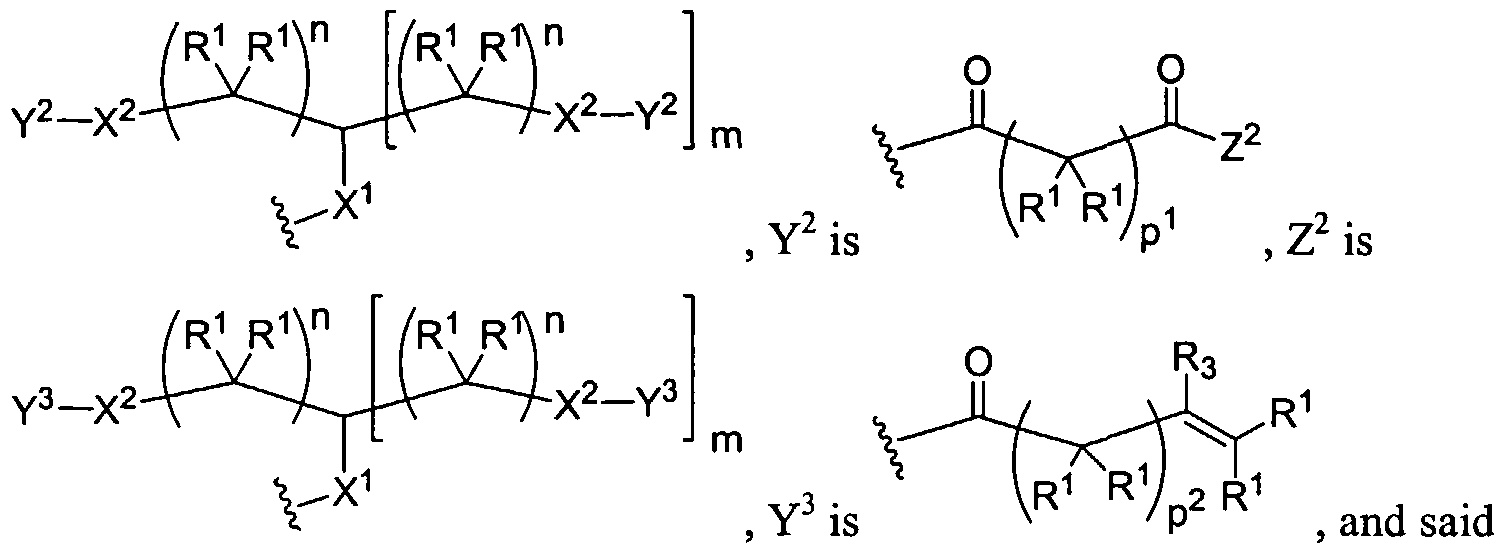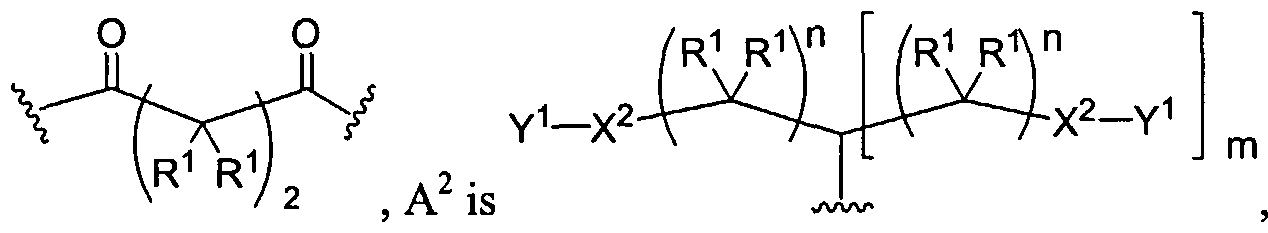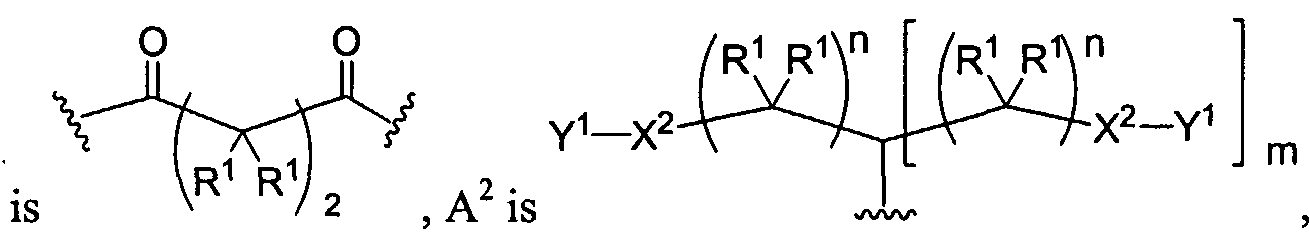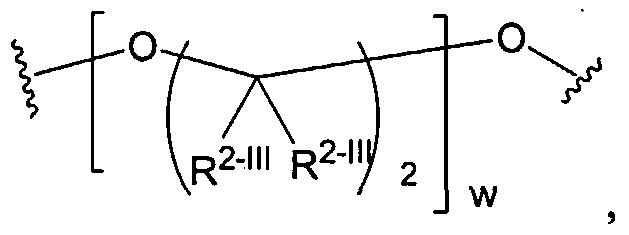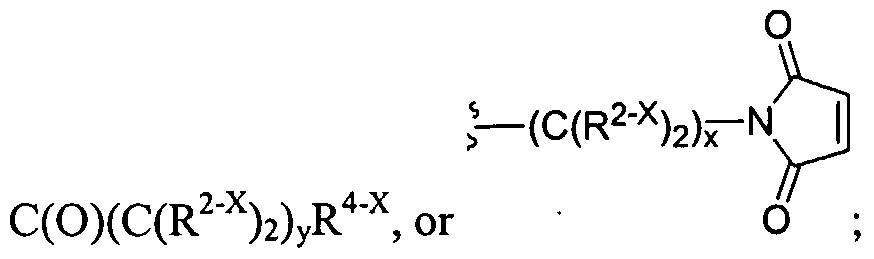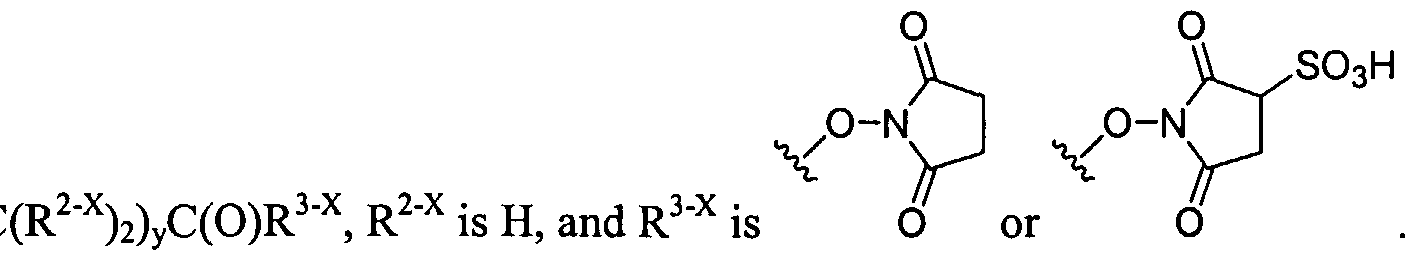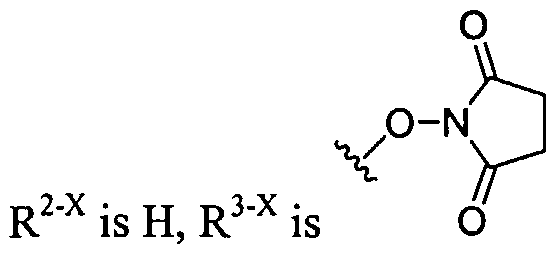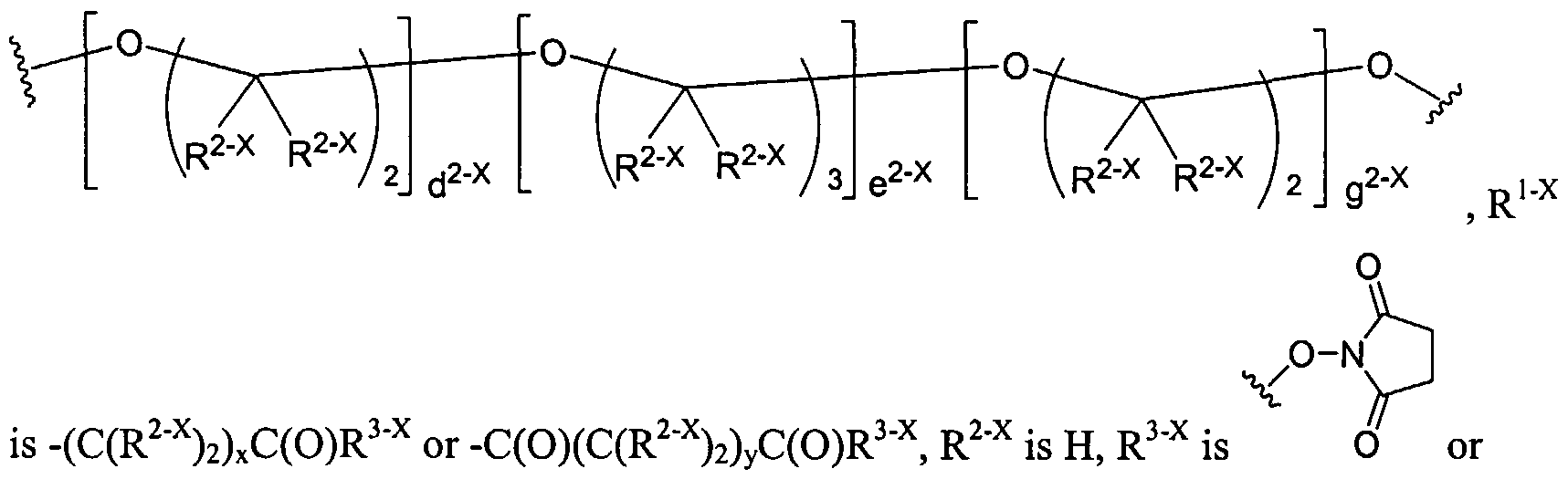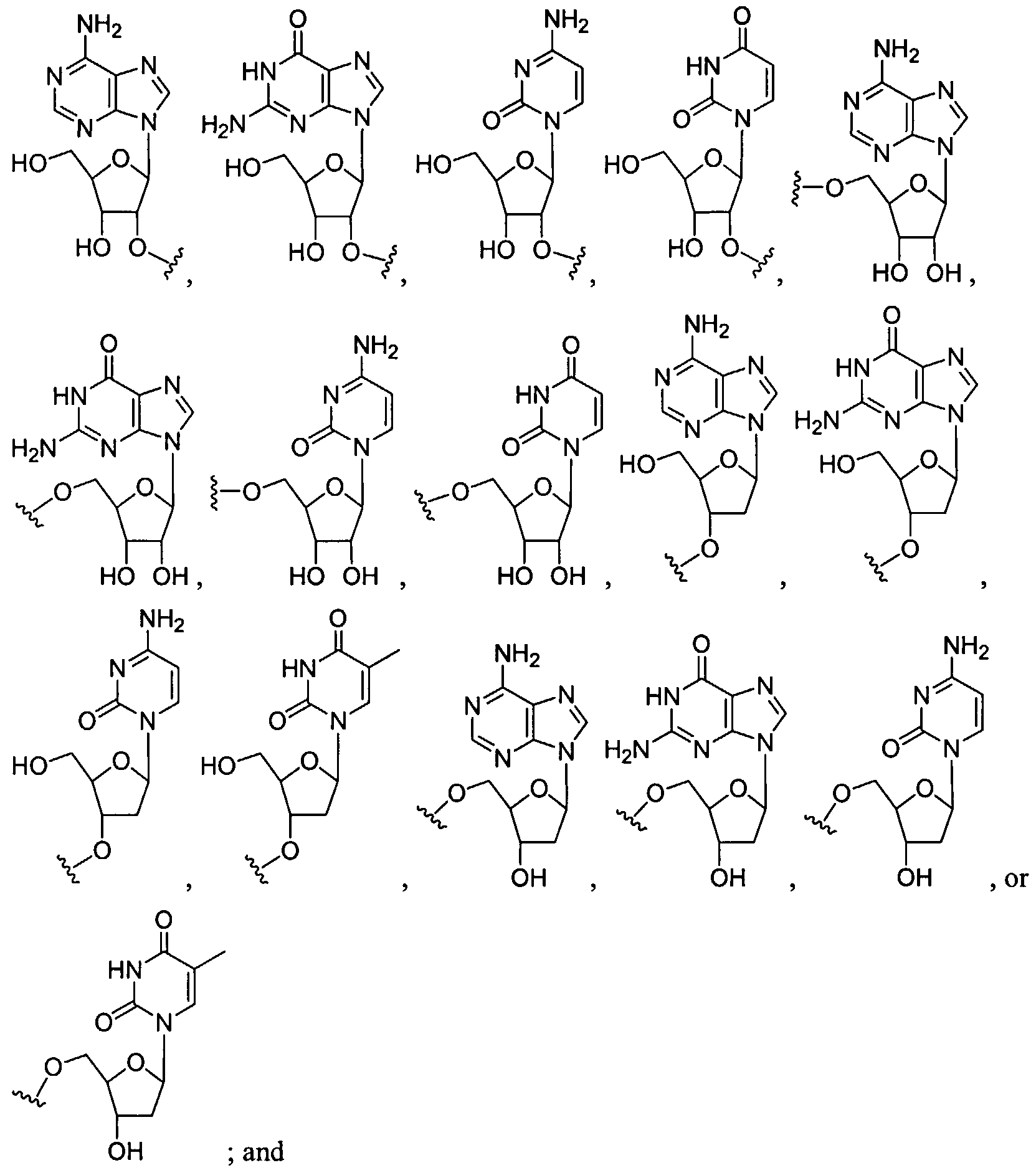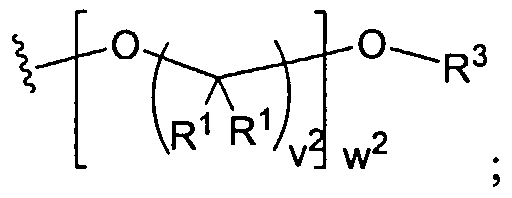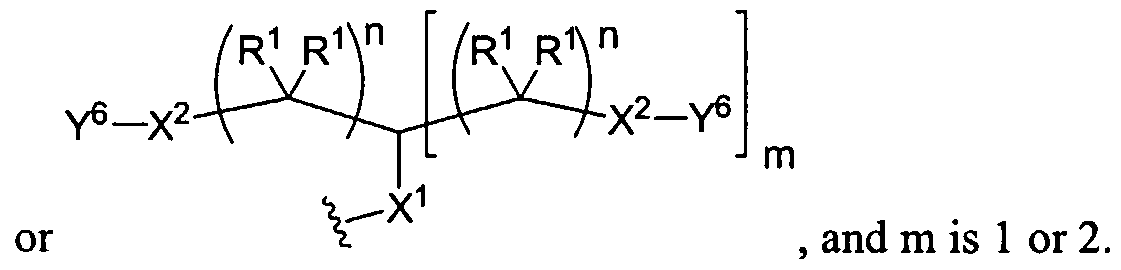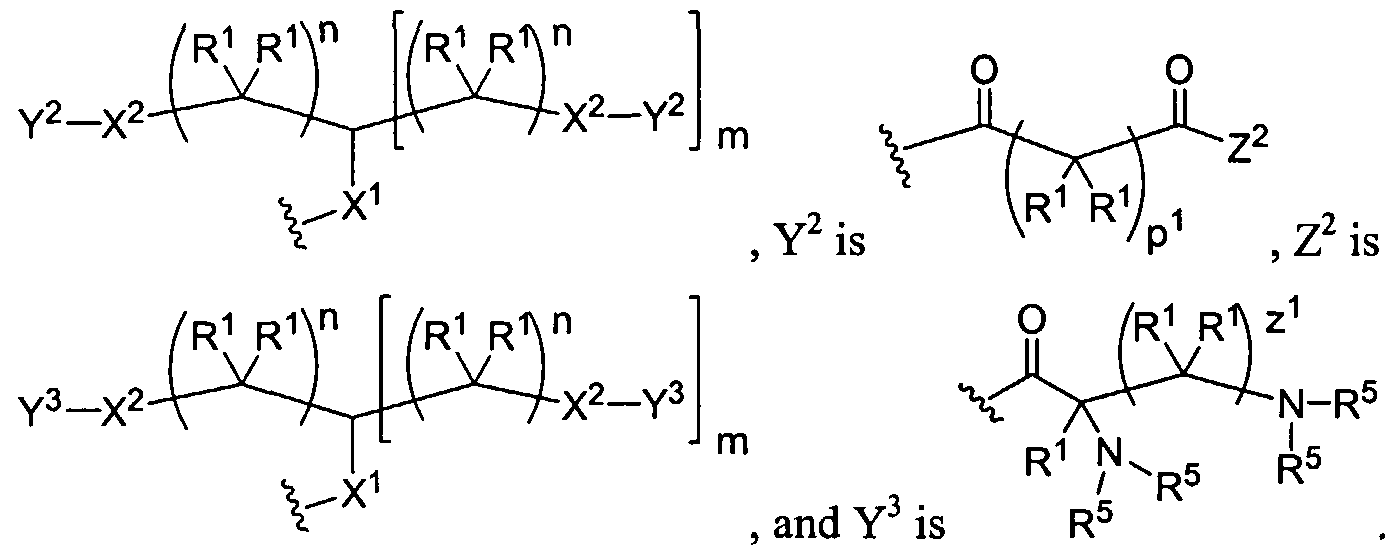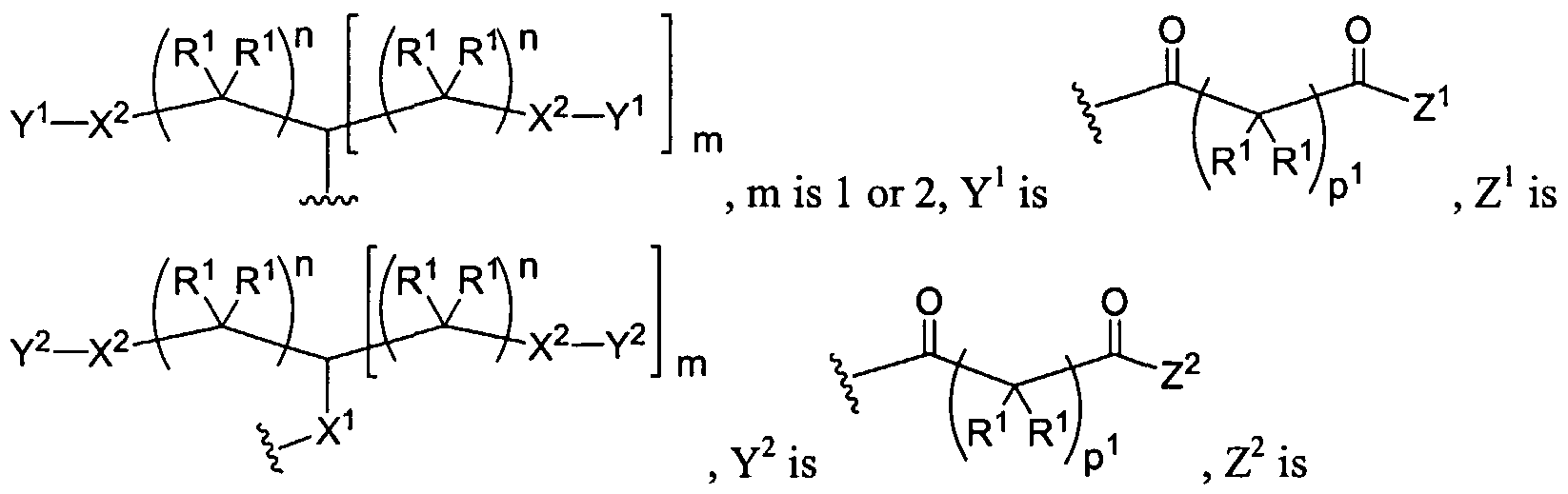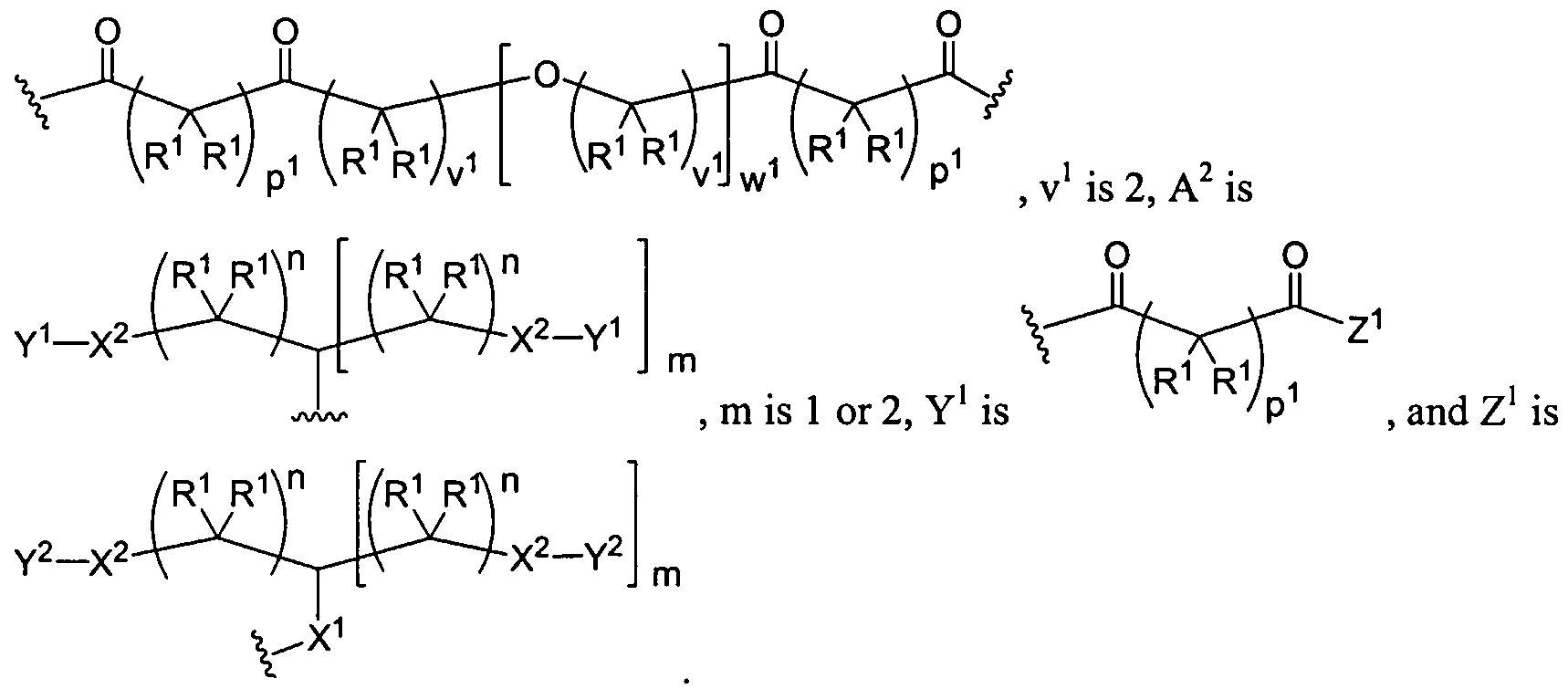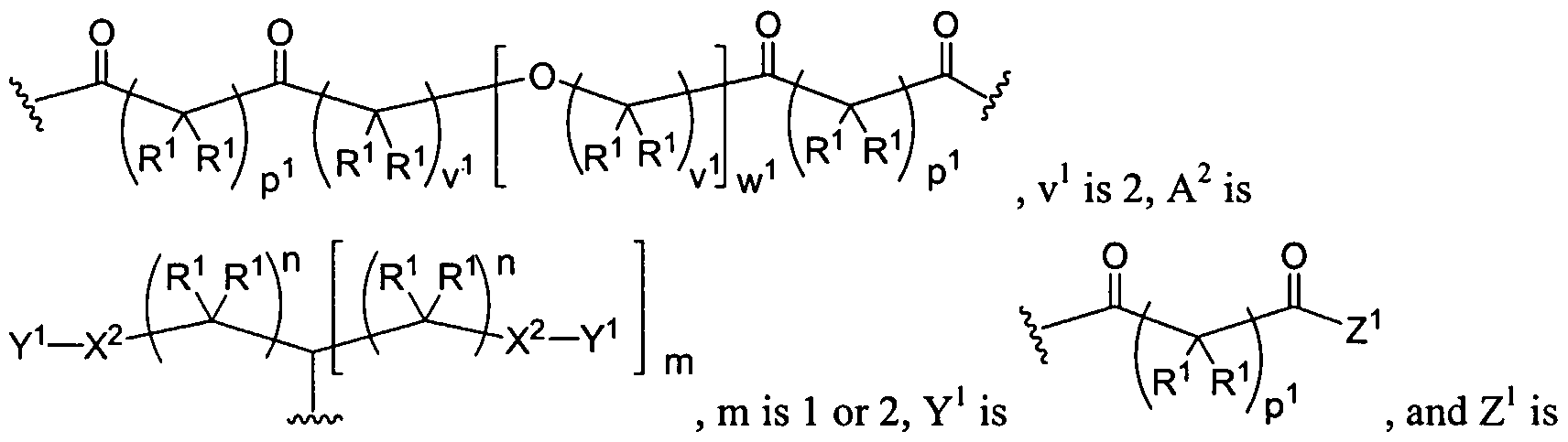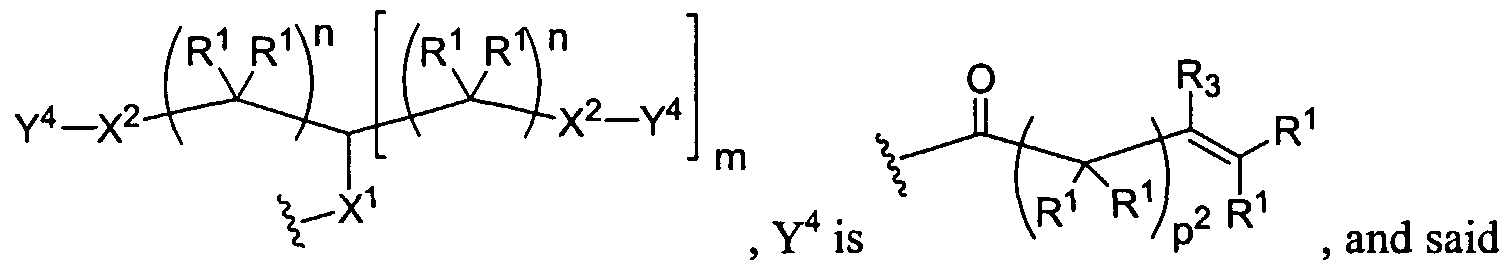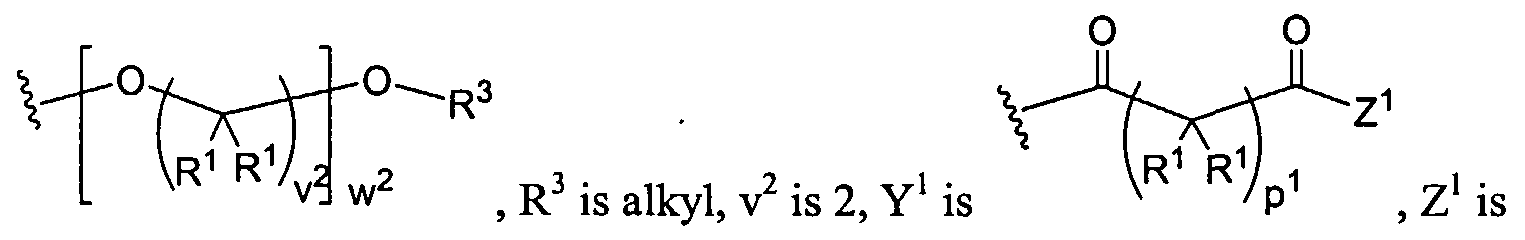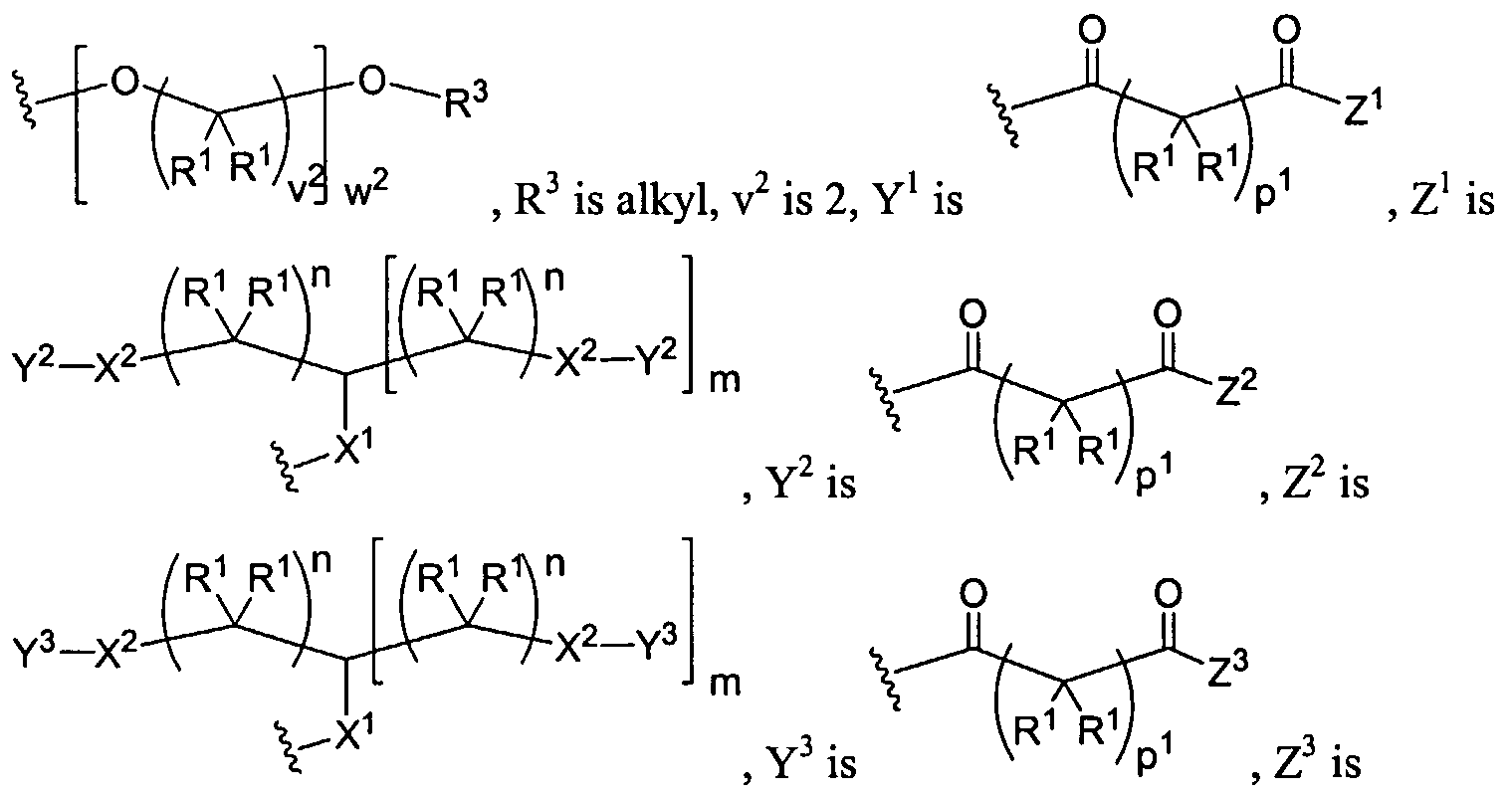Dendritic Polymers, Crosslinked Gels, and Their Uses in Orthopedic Applications
Related Applications
This application claims the benefit of priority to United States Provisional Patent Application serial number 60/603,502, filed August 20, 2004; and United States Provisional Patent Application serial number 60/604,097, filed August 23, 2004; both of which are incorporated by reference.
Background of the Invention
Cartilaginous tissues play important roles in contributing to load support and energy dissipation in the joints of the musculoskeletal system. These tissues include articular cartilage which is predominantly an avascular and alymphatic tissue with very low cell- density. As a result, articular cartilage has limited capacity for self-repair following injury or aging. Degeneration of cartilage in the meniscus, interverebral disks, or joints can lead to severe and debilitating pain in patients. Injuries to these tissues are often retained for many years and may eventually lead to more severe secondary damage. See Moskowitz, R. W., Osteoarthritis: diagnosis and medical/surgical management. 2" ed.; W.B. Saunders Company: 1984. Cuurently, more than one million knee, hip, and shoulder joint surgical procedures are performed annually in the United States as a consequence of trauma or a lifetime of wear and tear. See Praemer, A.; Furner, S.; Rice, D. P. Musculoskeletal Conditions in the United States, American Academy of Orthopaedic Surgeons: Rosemont, Illinois, 1999. Despite the large number of patients suffering from cartilage degeneration, the only widely-available treatment options for cartilage degeneration are chronic administration of anti-inflammatory agents, total joint replacement, osteotomy, or allograft transplantation, each of which leads to mixed long-term results. The primary functions of articular cartilage and the meniscus are to minimize contact stresses and contribute to energy dissipation and lubrication mechanisms during joint loading. Mow, V. C; Ratcliffe, A. Structure and Function of Articular Cartilage and Meniscus. 2nd ed.; Lippincott Raven: Philadelphia, 1997. These cartilaginous tissues behave as fiber-reinforced composites that are saturated with water (65-85% of tissue
weight). Deformation and loading of the tissues gives rise to large interstitial fluid pressures that are important for distributing load, reducing matrix stresses and strains, and dissipating energy. Negative charges associated with tissue proteoglycans confer hydrophilicity and a propensity to swell that is important for maintaining tissue hydration and recovery of initial geometry after loading. The nearly frictionless surface of articular cartilage is unmatched in vivo and provides for important sliding articulations in the shoulder, hip, and knee. In addition to the important load distribution role of the meniscus, the meniscus contributes to overall joint stability which may be compromised with injury. See Levy, I. M.; Torzilli, P. A.; Fisch, I. D. The contribution of the menisci to the stability of the knee. In Knee Meniscus. Basic and Clinical Foundations; Mow, V. C; Arnoczky, S. P.; Jackson, D. W., Eds.; Raven Press: New York, 1992; pp 107-116.
Articular cartilage and meniscus contain collagen fibers that are highly, and variably, oriented through the tissue. See Heinegard, D.; Bayliss, M. J.; Lorenzo, P. Biochemistry and metabolism of normal and osteoarthritic articular cartilage. In Osteoarthritis; Brandt, K. D.; Doherty, M.; Lohmander, L. S., Eds.; Oxford University Press: New York, 1998; pp 74-84. The collagen fibril network in articular cartilage consists principally of type II collagen (a defining characteristic), while the meniscus mostly contains large diameter and circumferentially oriented type I collagen fibers. Both matrices are distributed with smaller amounts of other collagens (i.e., types III, V, and VI for meniscus; types XI, VI, and IX for articular cartilage) and negatively-charged proteoglycans including aggrecan, decorin and biglycan. See Adams, M. E.; Hukins, D. W. L. The extracellular matrix of the meniscus. In Knee meniscus: Basic and Clinical Foundations; Mow VC, A. S., Jackson DW, Eds.; Raven Press: New York, 1992; pp 15-28 and Eyre, D. Collagen structure and function in articular cartilage. In Osteoarthritic Disorders; Kuettner, K.; Goldberg, V., Eds.; American Academy of Orthopaedic Surgeons: Rosemont, IL, 1995; pp 219-228. Cells of articular cartilage, the chondrocytes, are heterogeneous in their biosynthetic activity and morphology. See Buschmann, M. D.; Gluzband, Y. A.; Grodzinsky, A.; Kimura, J. H.; Hunziker, E. B. Journal of Orthopaedic Research 1992, 10, 745-758. Similarly, the meniscus contains complex subpopulations of cells that exhibit both fibroblast-like and chondrocyte-like morphologies and biological activities that may vary with region. Webber, R. J.; Harris, M. G.; Hough, A. J. J. Orthop.
Res. 1985, 3, 36-42. For these and all cartilage, native cells are responsible for both the synthesis and maintenance of the composition and structure of the extracellular matrix over the lifetime of a joint.
In a healthy joint, articular cartilage is able to withstand the large forces associated with load-bearing and joint motion over the lifetime of an individual. Following injury or with osteoarthritis, cartilage may exhibit fibrillation or cracking of the articular surface with partial or complete loss of the tissue. See Pritzker, K. P. H., Pathology of osteoarthritis. In Osteoarthritis; Brandt, K. D.; Doherty, M.; Lohmander, L. S., Eds.; Oxford University Press: New York, 1998; pp 50-62. Articular cartilage is limited in its ability to self-repair due to the lack of a vascular supply. Most surgical strategies commonly used for cartilage repair involve techniques that promote vascularization or cellular infiltration of the damaged cartilage in an attempt to promote a spontaneous repair response that would otherwise not be possible (e.g., osteochondral grafting, drilling, chondroplasty, and cell transplantation). See Brittberg, M.; Lindahl, A.; Nilsson, A.; Ohlsson, C; Isaksson, O.; Peterson, L. New England Journal of Medicine 1994, 331(14), 889-895; Buckwalter, J. A.; Mankin, H. J. Arthritis and Rheumatism 1998, 41(8), 1331-1342; and Hunziker, E. B. Osteoarthritis and Cartilage 2002, 10(6), 432-463. These traditional approaches have had varied success.
Damage to the meniscus following trauma or injury may lead to degenerative joint changes such as osteophyte formation, articular cartilage degeneration, joint space narrowing, and symptomatic osteoarthritis. See Fairbank, T. J. Journal of Bone and Joint
Surgery 1948, 3OB, 664-670 and Roos, H.; Adalberth, T.; Dahlberg, L.; Lohmander, L. S.
Osteoarthritis and Cartilage 1995, 3(4), 261-267. Although partial or total meniscectomy may be performed for a range of injuries from small radial tears to more severe and complex tears, restoration of normal meniscal function is the treatment of choice when technically possible. The ability of a meniscal lesion to heal, either spontaneously or following surgical repair, is dependent on the proximity of the tear to the peripheral vascular supply, the size and complexity of the tear, and the integrity of the extracellular matrix. See Arnoczky, S. P.; Warren, R. F. Am. J. Sports Med. 1983, 11(3), 131-141. Longitudinal or circumferential lesions located within the peripheral 10-25% of the meniscus generally heal through formation of a fibrin clot that promotes the penetration of
blood vessels and undifferentiated mesenchymal cells and fibroblasts that form a fϊbrovascular scar in the lesion. See Henning, C. E.; Lynch, M. A.; Yearout, K. M.; Vequist, S. W.; Stallbaumer, R. J.; Decker, K. A. Clinical Orthopaedics & Related Research 1990, 252, 64-72. This spontaneous healing response is often associated with a long-term positive outcome. Tears that do not communicate with the vascular supply, such as radial tears or complex tears that span the avascular meniscal regions, may exhibit poor or no spontaneous healing and may benefit from surgical intervention.
In addition to the important contribution of autograft and allograft approaches to cartilage repair, synthetic and natural polymers have played important roles as cartilage repair scaffolds. A number of synthetic linear polymers have been implicatd in tissue engineering and related medical applications. See Christenson, L.; Mikos, A. G.; Gibbons, D. F.; Picciolo, G. L. Tissue Eng. 1997, 3, 71-76; Langer, R., Whitaker Annals of Biomedical Engineering 1995, 23(2), 101-11; and Lu, L. C; Zhu, X.; Valenzuela, R. G.; Currier, B. L.; Yaszemski, M. J. Clinical Orthopaedics and Related Research 2001, 391, S251-S270. Representative synthetic linear polymers include polyethylene glycol (PEG), poly(glycolic acid) (PGA), poly(lactic acid) (PLA), poly(caprolactone), poly(propylene fumarate), poly(NIPAMM), polyurethanes and various co-polymers. Bryant, S. J.; Anseth, K. S. J. Biomed. Mater. Res. 2002, 59, 63-72; Elisseeff, J.; Mclntosh, W.; Anseth, K. S.; Riley, S.; Ragan, P.; Langer, R. J. Biomed. Mat. Res 2000, 51, 164-171; Chu, C. R. et al. J. Biomed. Mat. Res. 1995, 29(9), 1147-54; Freed, L. E.; Marquis, J. C; Nohria, A.; Emmanual, J.; Mikos, A. G.; Langer, R. J. Biomed. Mat. Res. 1993, 27(1), 11-23; Ibusuki, S. et al. Tissue Eng 2003, 9(2), 371-84; Ibusuki, S. et al. A. Tissue Eng 2003, 9(6), 1133- 42; Temenoff, J. S.; Mikos, A. G. Biomaterials 2000, 21(23), 2405-2412; Cao, T.; Ho, K. H.; Teoh, S. H., Tissue Engineering 2003, 9, S103-S112; Moran, J. M.; Pazzano, D.; Bonassar, L. J., Tissue Engineering 2003, 9 (1), 63-70; Schaefer, D.; Martin, L; Jundt, G.; Seidel, J.; Heberer, M.; Grodzinsky, A.; Bergin, I.; Vunjak-Novakovic, G., Arthritis & Rheumatism 2002, 46 (9), 2524-2534; and Freed, L. E.; Grande, D. A.; Lingbin, Z.; Emmanual, J.; Marquis, J. C; Langer, R., J. Biomed. Mater. Res. 1994, 28 (8), 891-899.
Natural scaffolds have also been widely studied for cartilage repair, including alginate, agarose, hyaluronan, chitosan, fibrin, type I and II collagen, and small intestine submucosa. For example, see Grigolo, B.; Roseti, L.; Fiorini, M.; Fini, M.; Giavaresi, G.;
Aldini, N. N.; Giardino, R.; Facchini, A. Biomaterials 2001, 22(17), 2417-24; Nehrer, S. et al. J. Biomed. Mat. Res. 1997, 38(2), 95-104; Paige, K. T.; Cima, L. G.; Yaremchuk, M. J.; Vacanti, J. P.; Vacanti, C. A. Plastic Reconstruct. Surgery 1995, 96(6), 1399-1400; Solchaga, L. A.; Dennis, J. E.; Goldberg, V. M.; Caplan, A. I. J. Ortho. Res. 1999, 17(2), 205-13; and Wakitani, S. et al. Tissue Eng. 1998, 4(4), 429-444;- Lee, C. R.; Grodzinsky, A. J.; Hsu, H. P.; Spector, M., J. Orthop. Res. 2003, 21 (2), 272-281; Klein, T. J.; Schumacher, B. L.; Schmidt, T. A.; Li, K. W.; Voegtline, M. S.; Masuda, K.; Thonar, E. J. M. A.; Sah, R. L., Osteoarthr. Cartilage 2003, 11 (8), 595-602; van Susante, J. L. C; Pieper, J.; Buma, P.; van Kuppevelt, T. H.; van Beuningen, H.; van der Kraan, P. M.; Veerkamp, J. H.; van den Berg, W. B.; Veth, R. P. H., Biomaterials 2001, 22 (17), 2359- 2369; Chenite, A.; Chaput, C; Wang, D.; Combes, C; Buschmann, M. D.; Hoemann, C. D.; Leroux, J. C; Atkinson, B. L.; Binette, F.; Selmani, A., Biomaterials 2000, 21 (21), 2155-2161; Silverman, R. P.; Bonasser, L.; Passaretti, D.; Randolph, M. A.; Yaremchuk, M. J., Plast. Reconstruct. Surg. 2000, 105 (4), 1393-1398; Brun, P.; Cortivo, R.; Zavan, B.; Vecchiato, N.; Abatangelo, G., J. Mater. Sci.-Mater. Med. 1999, 10 (10-11), 683-688; Brun, P.; Abatangelo, G.; Radice, M.; Zacchi, V.; Guidolin, D.; Daga Gordini, D.; Cortivo, R., J. Biomed. Mater. Res. 1999, 46 (3), 337-46; and Silverman, R. P.; Passaretti, D.; Huang, W.; Randolph, M. A.; Yaremchuk, M., Plast. Reconstruct. Surg. 1999, 103 (7), 1809-1818. There have been significant advances in recent years, with evidence that many of these materials promote cartilage growth in vitro following culture in soluble factors (e.g., dexamethasone or TGF-beta) under well-controlled bioreactor conditions. See Freed, L. E.; Hollander, A. P.; Martin, L; Barry, J. R.; Langer, R.; Vunjak-Novakovic, G. Exp. Cell Res. 1998, 240(1), 58-65; Mauck, R. L.; Soltz, M. A.; Wang, C. C. B.; Wong, D. D.; Chao, P. H. G.; Valhmu, W. B.; Hung, C. T.; Ateshian, G. A. Journal of Biomechanical Engineering-Transactions of the Asme 2000, 122(3), 252-260; van der Krann, P. M.; Buma, P.; van Kuppevelt, T.; van den Berg, W. B. Osteoarthritis and Cartilage 2002, 10(8), 631- 7; and Vunjak-Novakovic, G.; Obradovic, B.; Martin, I.; Freed, L. E. Biorheology 2002, 39(1-2), 259-268. Several cell-seeded scaffolds can approach the compressive stiffness and hydraulic permeability of native articular cartilage following culture under controlled bioreactor conditions. Some scaffolds have been evaluated in animal models of osteochondral defects with evidence of some "positive outcomes" as evaluated by
histological appearance, biochemistry, or immunohistochemistry. The overwhelming majority of the scaffolds studied for in vivo repair were formed in vitro and implanted into the tissue defect. The implantation generally used suture fixation and often resulted in problems integrating the implant with the adjacent tissue. Biomaterials that polymerize in situ are generally low-viscosity solutions that permit mixing with cells and/or bioactive factors which may be polymerized or crosslinked in situ. The polymerization or crosslinking is usually accomplished via chemical initiation or the use of light. Thus, these polymers may readily flow into and fill an irregularly shaped defect and be subsequently transformed from a liquid to a solid in a controlled manner. Scientific progress relating to in situ polymerization/solidification has advanced significantly in the last five years. For reviews see Hubbell, J. A. Mat. Res. Soc. Bull. 1996, 21, 33-35 and Nguyen, K. T.; West, J. L. Biomaterials 2002, 23, 4307-4314. In fact, a recent issue of Biomaterials (Biomaterials, vol. 23, 2002) was devoted entirely to the topic of Injectable Polymeric Biomaterials highlighting the importance of this research area and its potential rewards.
Alginate was one of the earliest materials to be investigated as an injectable in situ polymerizing scaffold for articular cartilage repair. More recently, investigators have proposed formulations for articular cartilage repair based on synthetic linear polymers that are either thermally or photochemically activated such as poly(N-isopropylacrylamide), poly(propylene fumarate), PEG, PVA, and poly(anhydrides). See Stile, R. A.; Burghardt, W. R.; Healy, K. E. Macromolecules 1999, 32(22), 7370-7379; Burkoth, A.; Anseth, K. Biomaterials 2000, 21, 2395-404; Schmedlen, R. H.; Masters, K. S.; West, J. L. Biomaterials 2002, 23, 4325-4332; and Temenoff, J. S.; Park, H.; Jabbari, E.; Conway, D. E.; Sheffield, T. L.; Ambrose, C. G.; Mikos, A. G., Biomacromolecules 2004, 5 (1), 5-10. A thermally responsive elastin-like polypeptide (ELP) scaffold for cartilage repair has been described as well as an injectable, photopolymerizing hyaluronan for cartilage repair. The hyaluronan-based hydrogels were evaluated in a rabbit osteochondral defect model with results that demonstrated good filling and integration with native tissue, cellular infiltration, and synthesis of a type II collagen-containing extracellular matrix. However, it is difficult to achieve optimal mechanical properties and optimal biochemical response with these linear polymers. For additional information see Smeds, K. A.; Wang, J. Y.; Baer, A. E.;
Setton, L. A.; Grinstaff, M. W. Transactions of the 2000 Annual Meeting of the Materials Research Society 2000, LLl.3, p. 207; Smeds, K.; Pfister-Serres, A.; Miki, D.; Dastghieb, K. A.; Inoue, M.; Hatchell, D. L.; Grinstaff, M. W. J. Biomed. Mat. Res. 2001, 54, 115-121; and Nettles, D. L.; Vail, T. P.; Morgan, M. T.; Grinstaff, M. W.; Setton, L. A. Ann. Biomed. Eng. 2004, in press.
Tissue engineering approaches promise the ability to repair or regenerate cartilaginous tissues by using combinations of cells, biomaterials, and biologically active molecules. Hence, the need exists for new materials and methods for repairing defects in cartilaginous tissue. The present invention fulfills this need, and has other related advantages.
Summary of the Invention
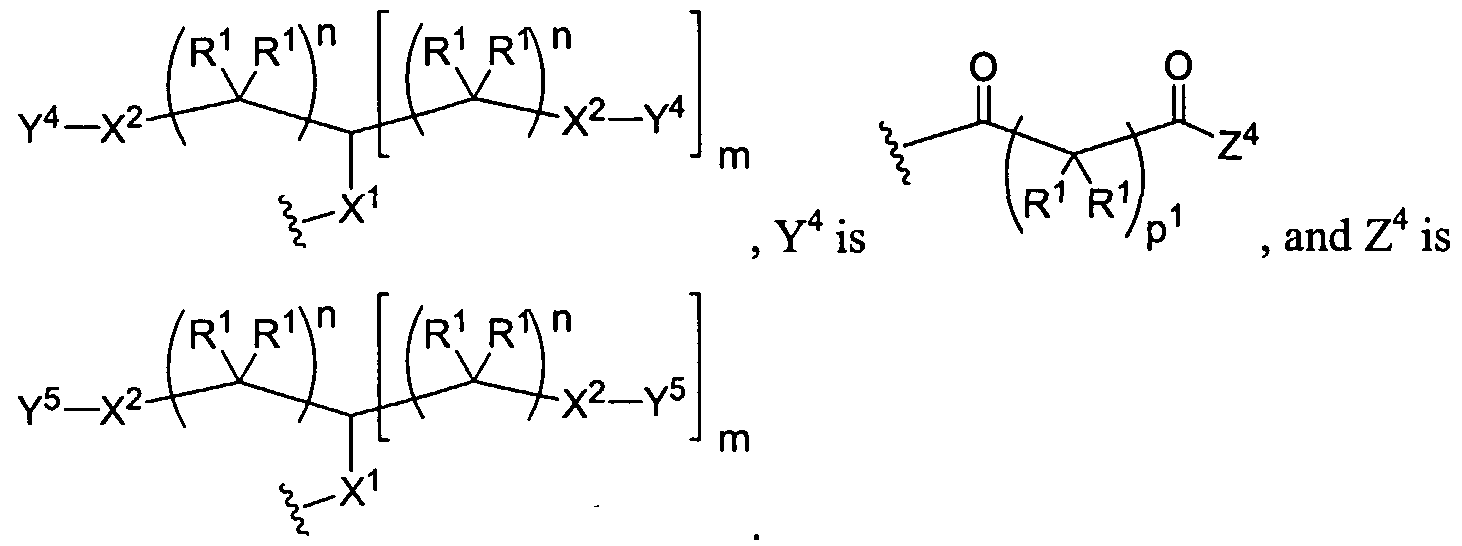
The present invention generally relates to methods of repairing defects in cartilaginous tissue. In certain instances, the cartilaginous tissue is articular. In certain instances, the compositions used to repair the tissue defect comprise a dendrimer. In certain instances, the dendritic polymers have an acrylate group attached at the periphery of the dendrimer. Treatment of the acrylate-capped dendritic polymers with ultraviolet radiation causes the dendritic polymers to polymerize forming a gel. In certain instances, the dendritic polymers have a lysine, cysteine, isocysteine residue or other nucleophilic group attached to the periphery of the dendrimer. Addition of a compound containing two or more electrophilic groups, such as aldehydes, activated esters, or acrylates, to the lysine- capped, cysteine-capped, or isocysteine-capped dendrimers produces a polymeric compound that can form a gel. In certain instances, the compositions used to repair the defect comprise a compound that has a polylysine core to which cysteine, isocysteine, or other nucleophilic groups are attached. Addition of a compound containing two or more electrophilic groups, such as aldehydes, activated esters, or acrylates, to the cysteine-capped or isocysteine-capped polylysine compounds produces a polymeric compound that can form a gel. In certain instances, the compound containing the electrophilic groups comprises a copolymer of polyethylene glycol and polypropylene glycol.
Brief Description of Figures
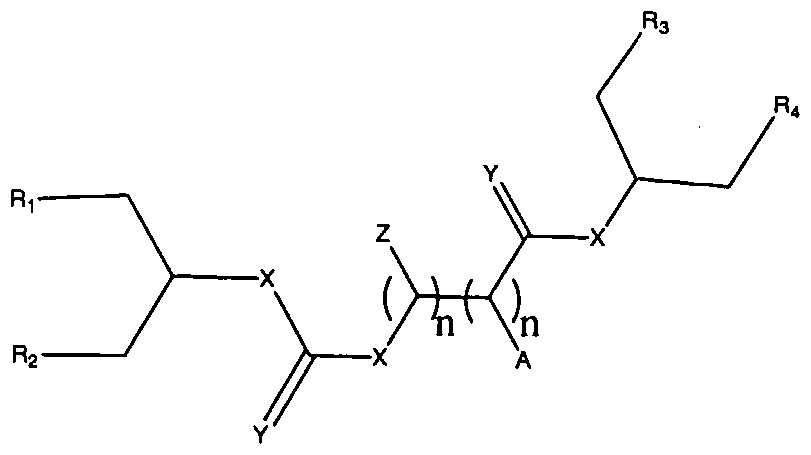
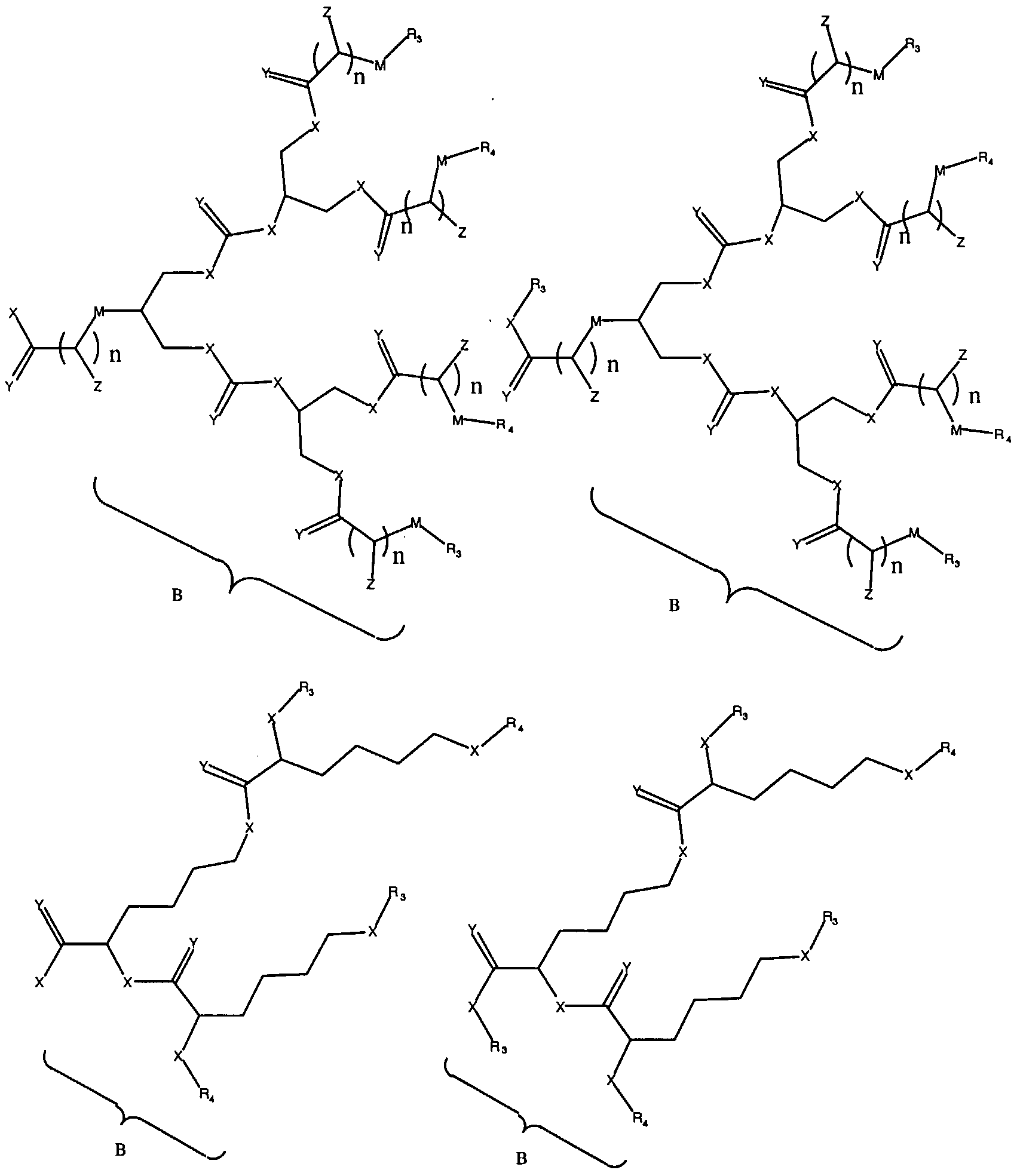
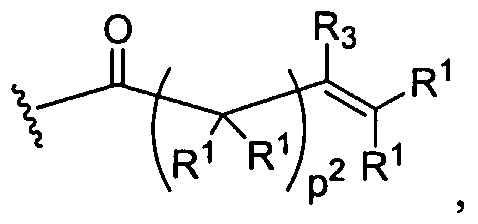
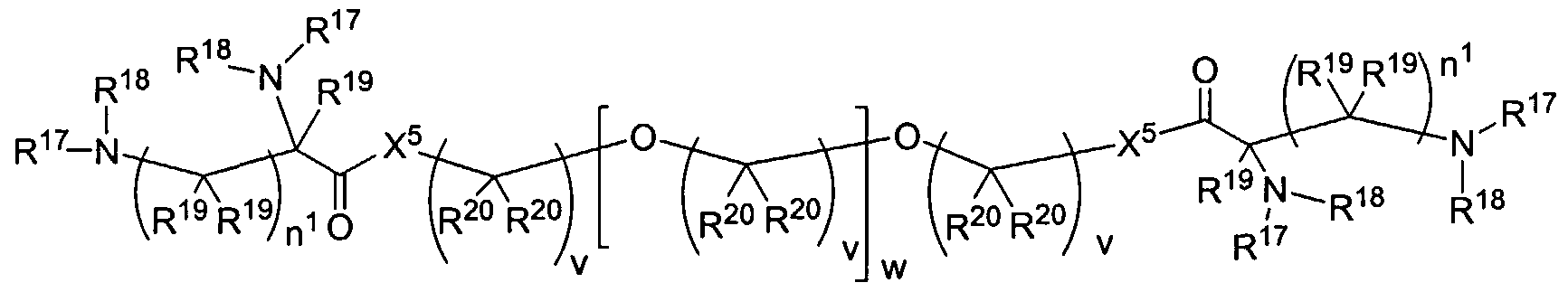
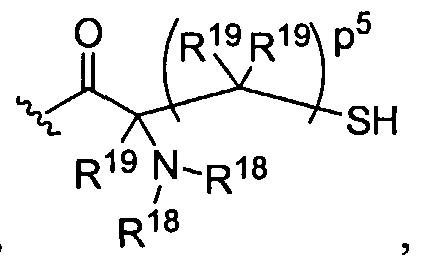
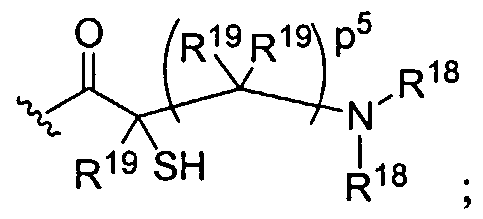
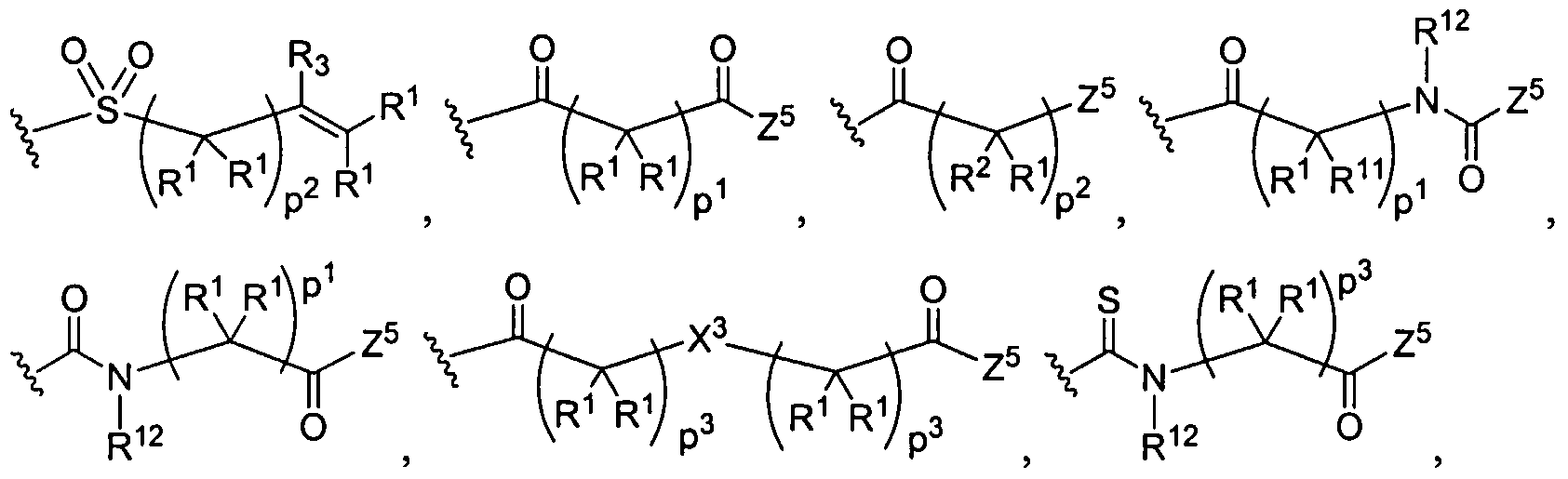
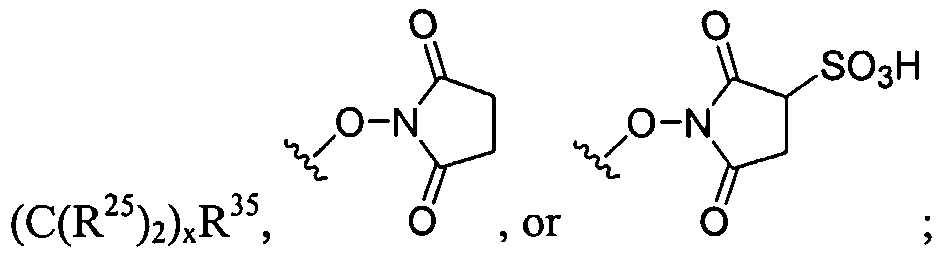
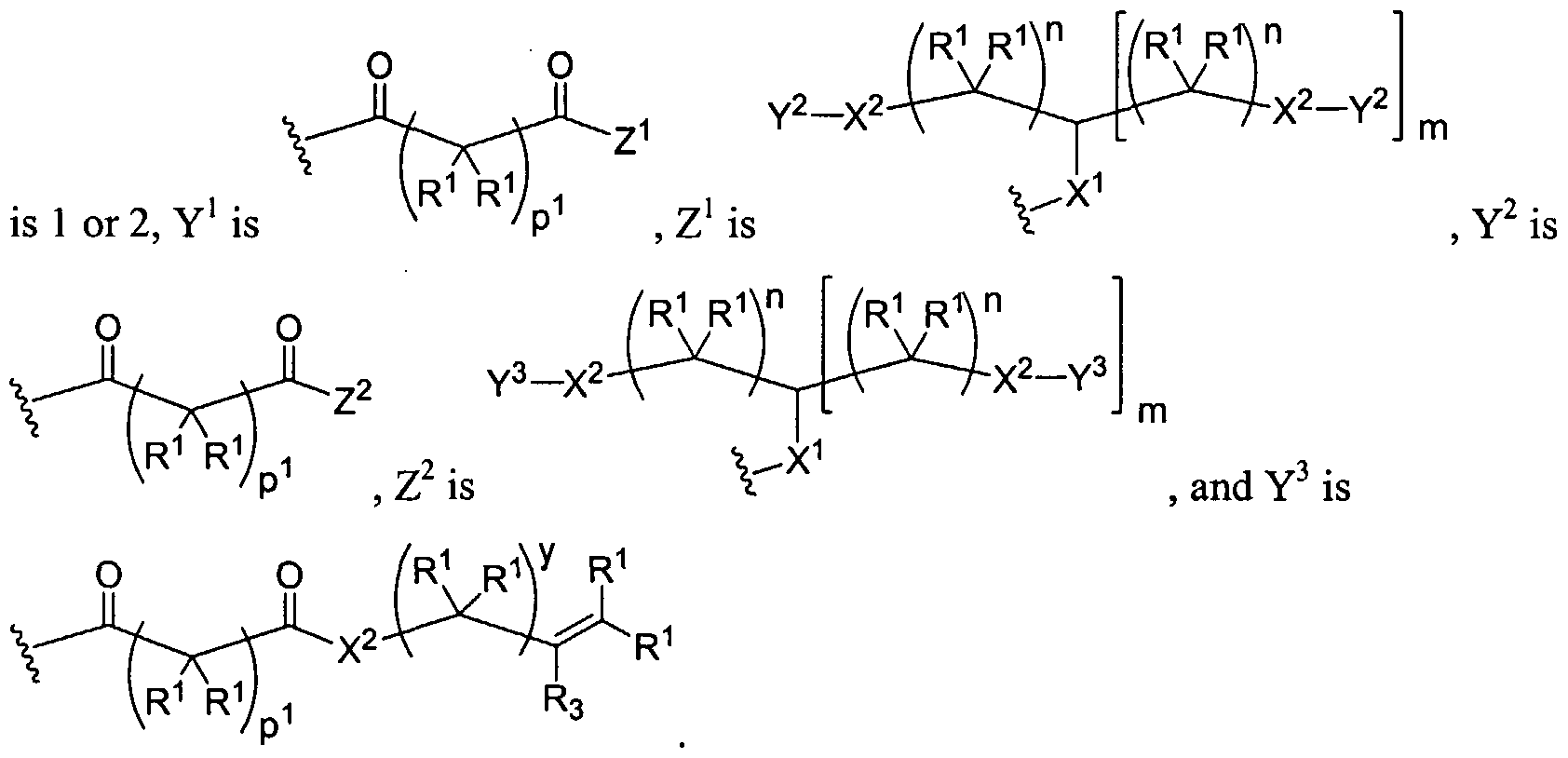
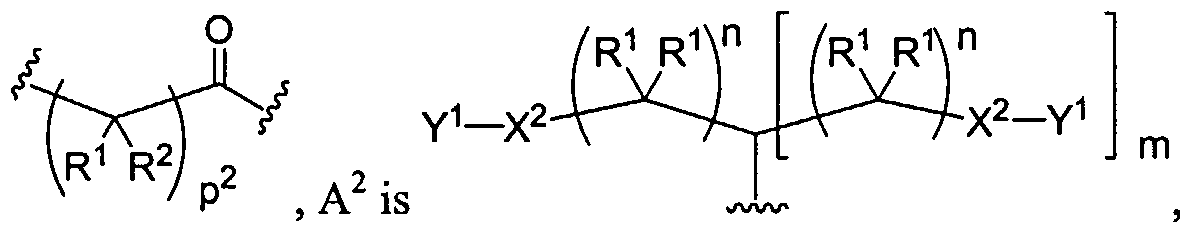
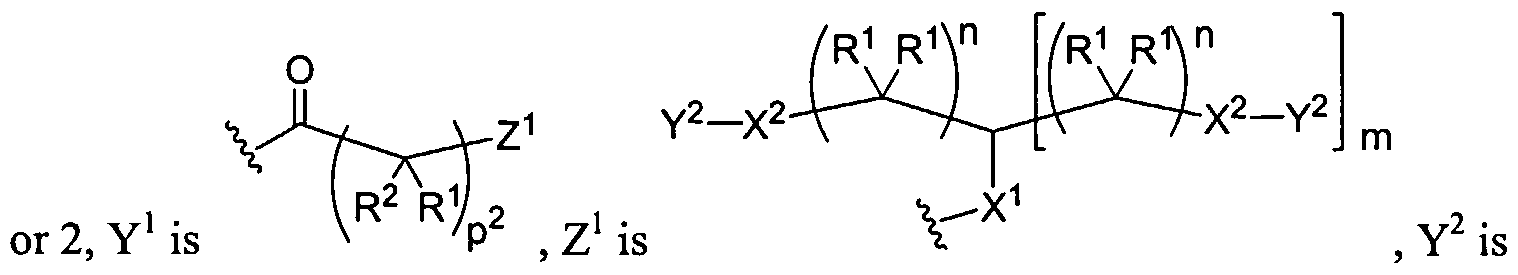
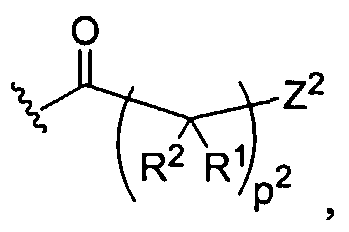
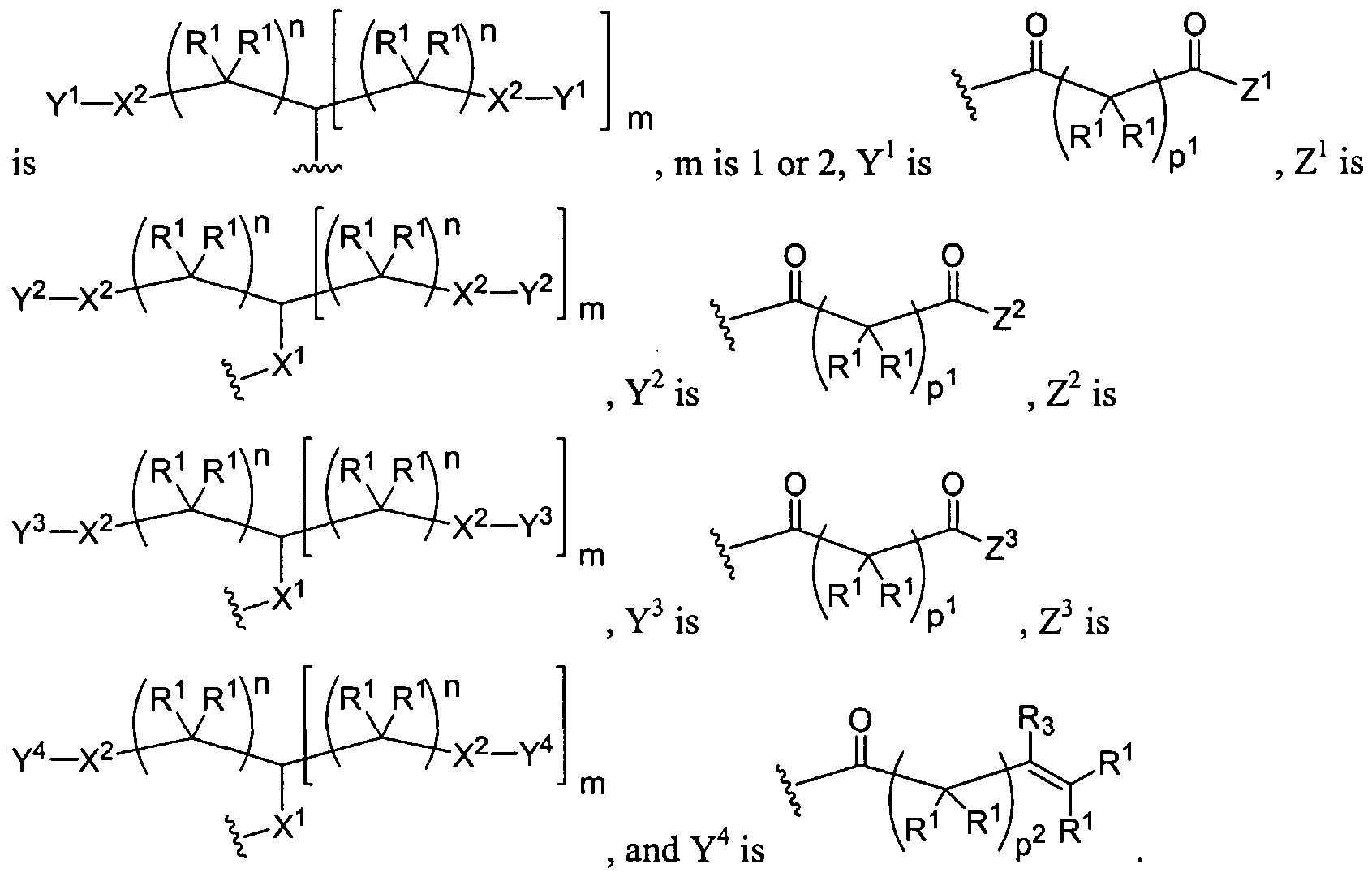
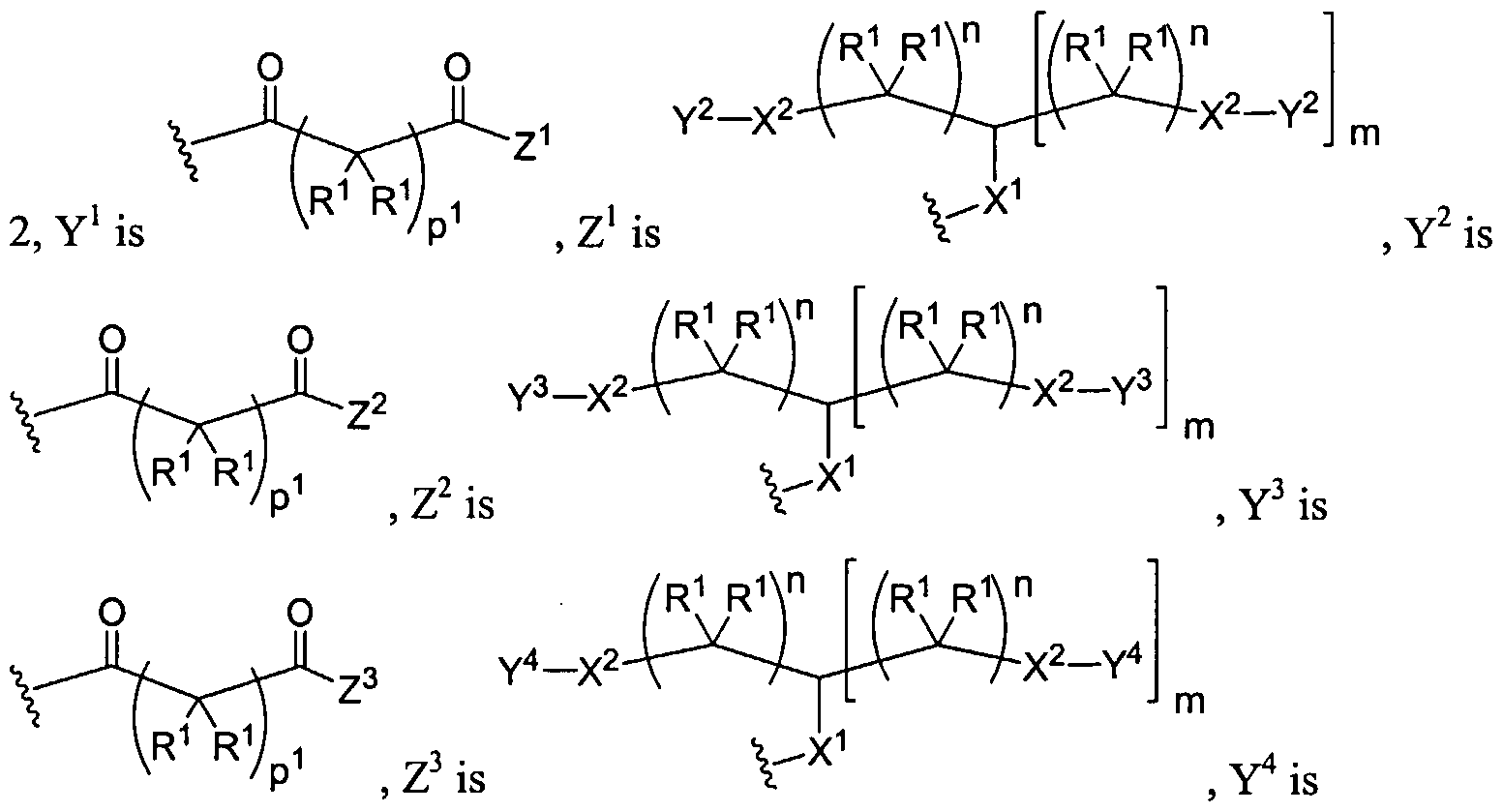
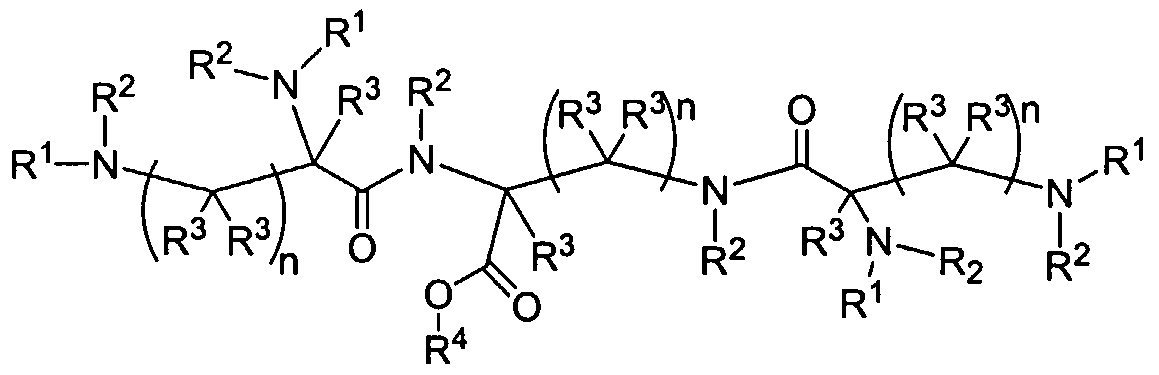
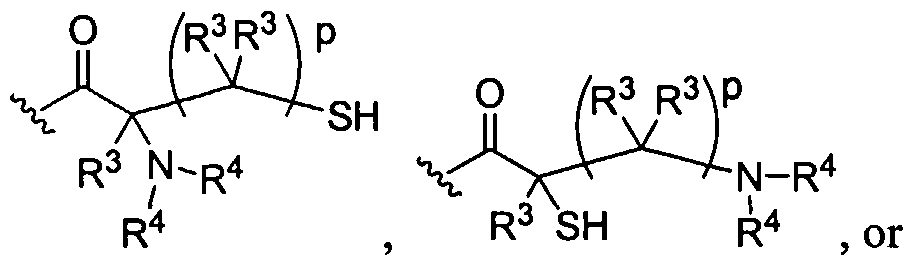
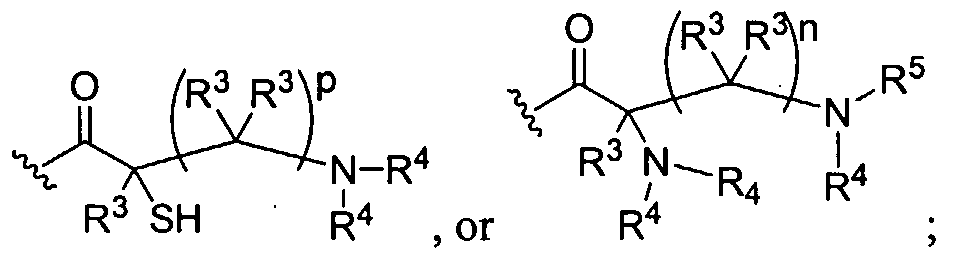
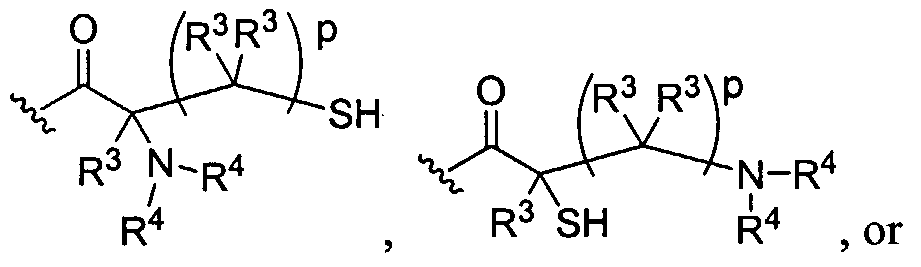
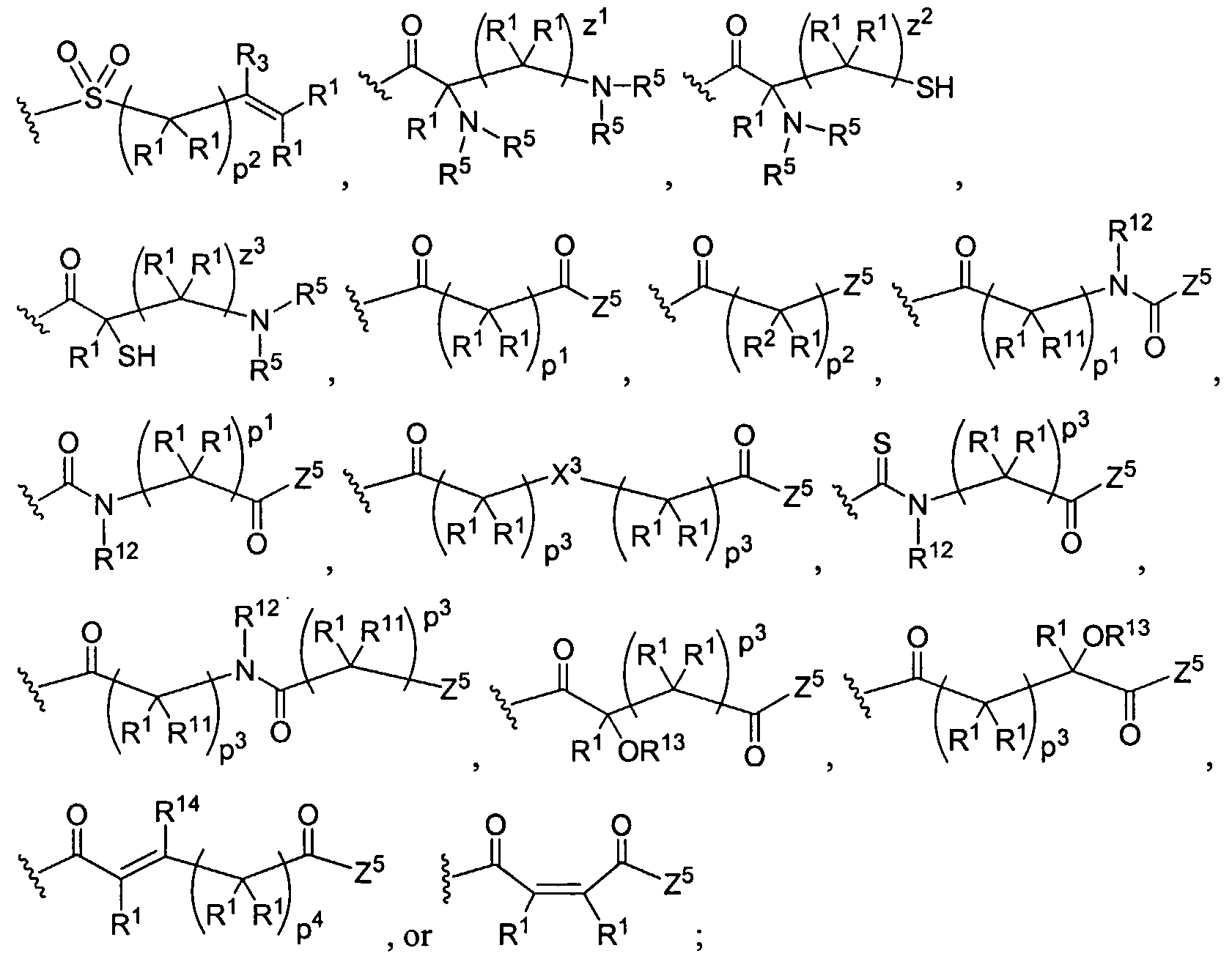
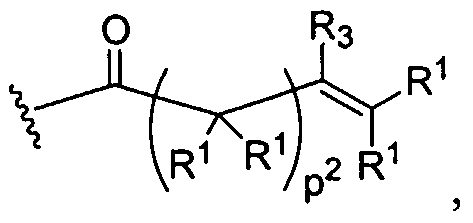
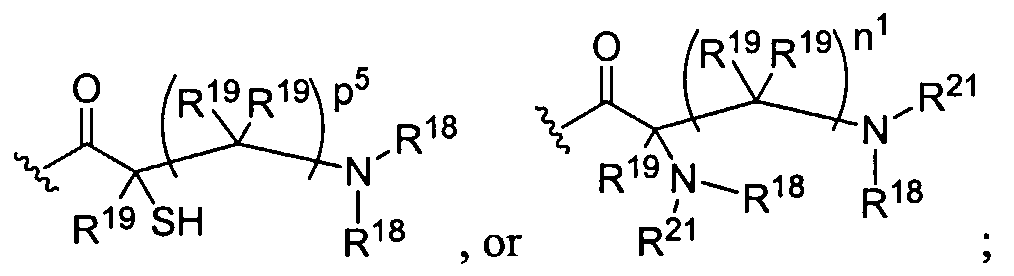
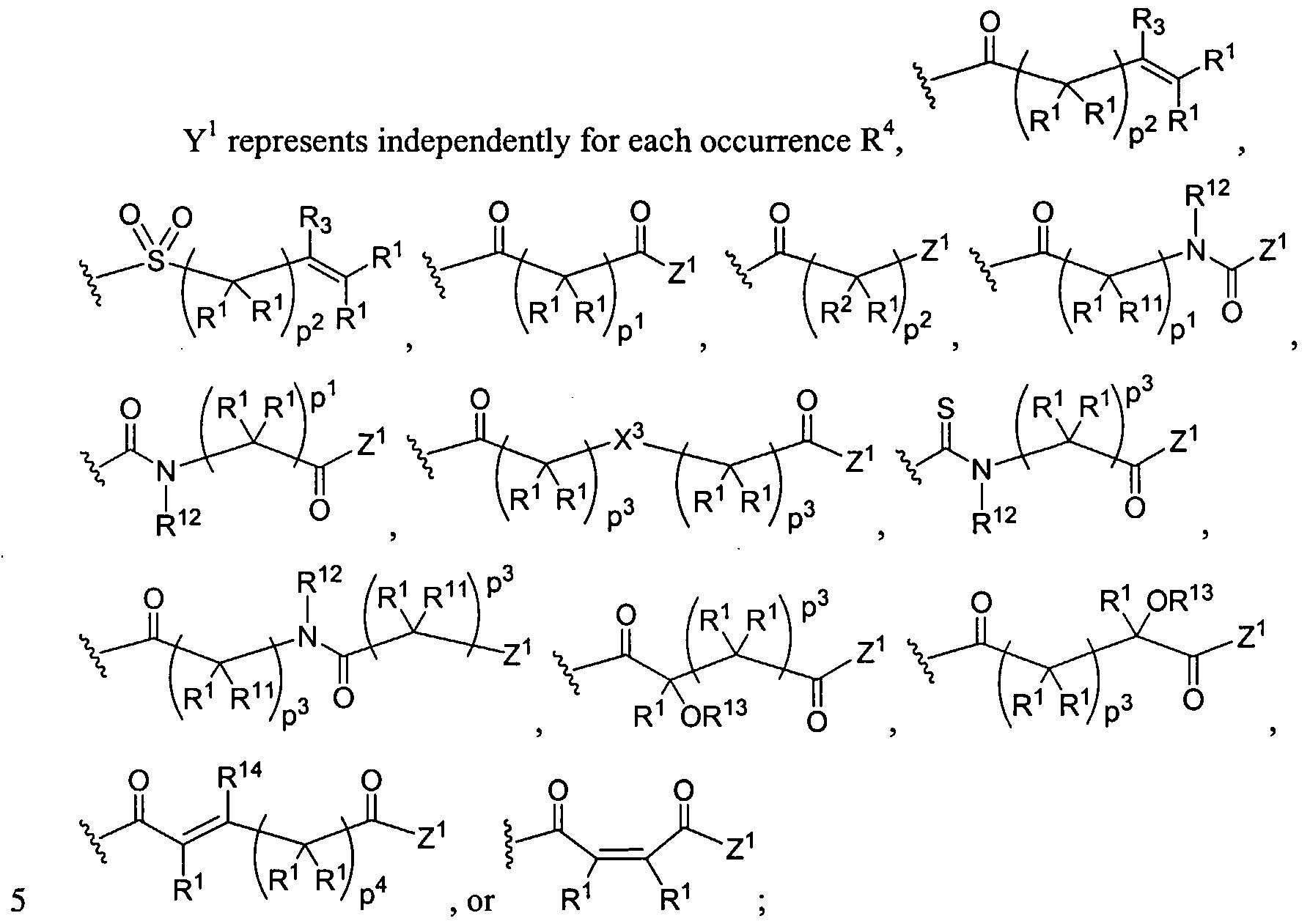
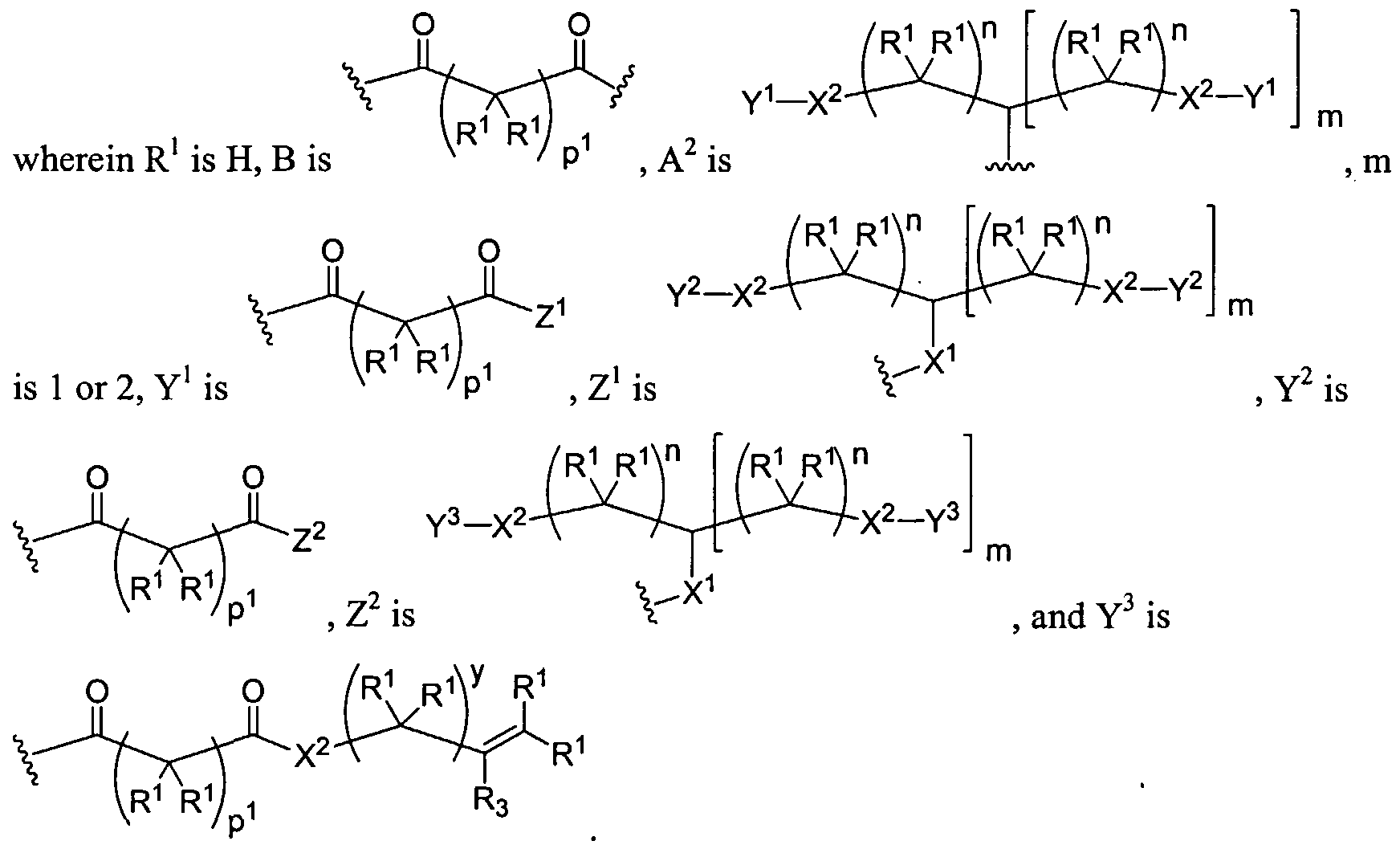
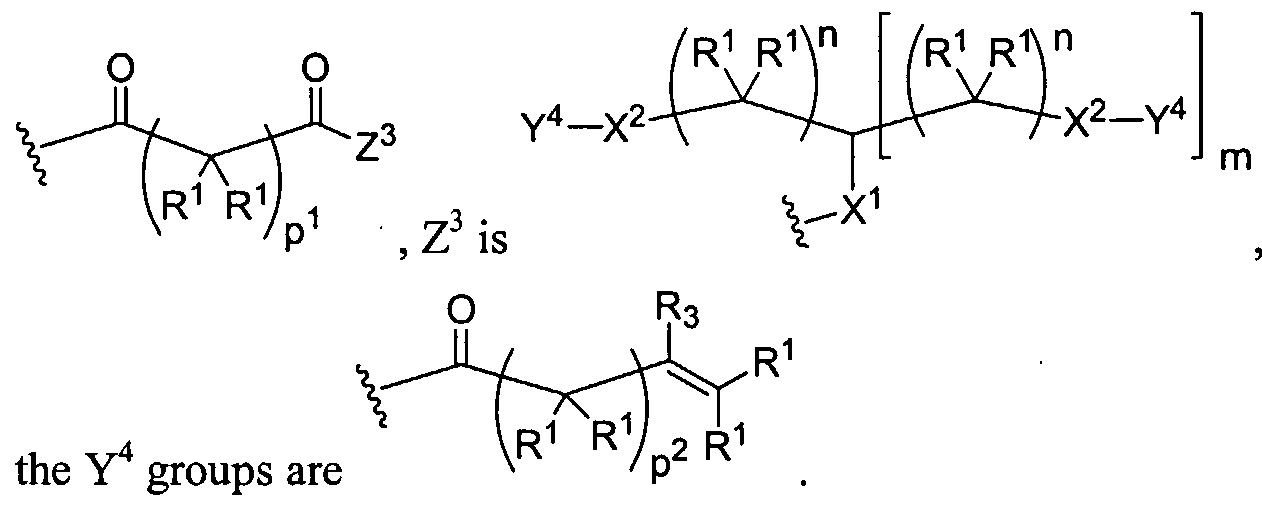
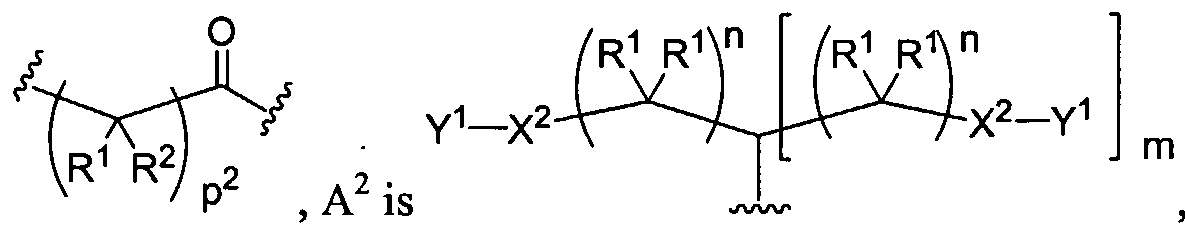
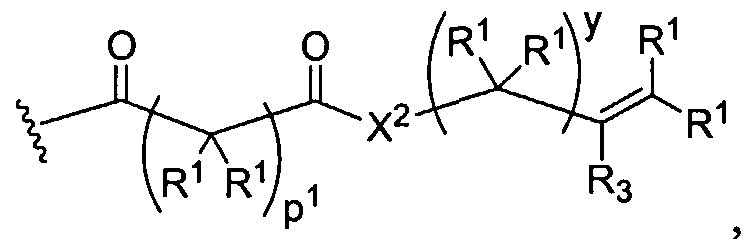
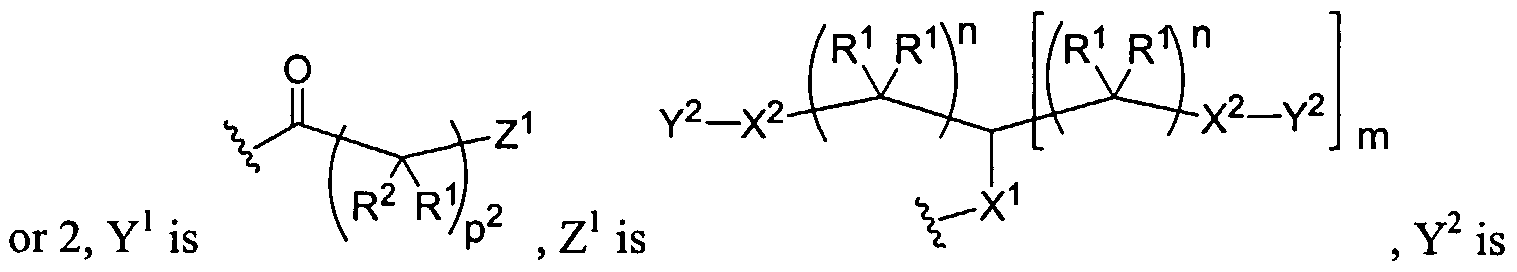
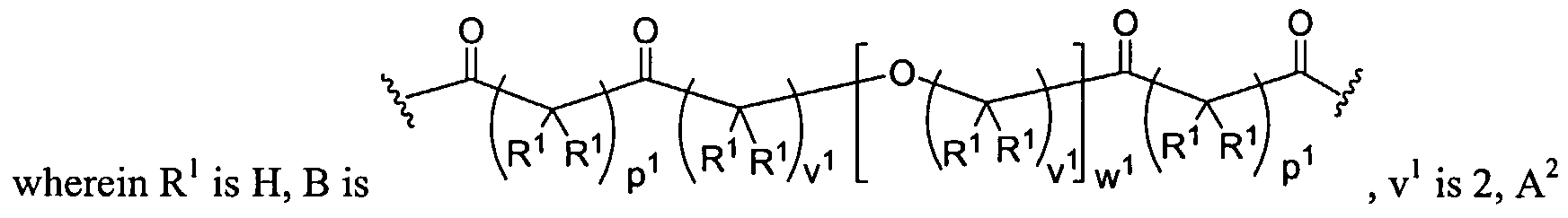
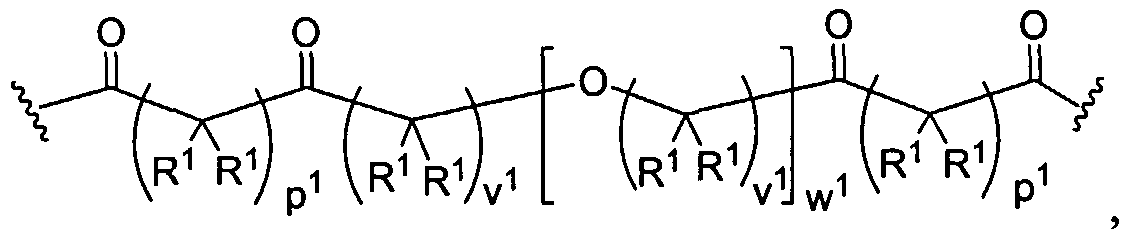
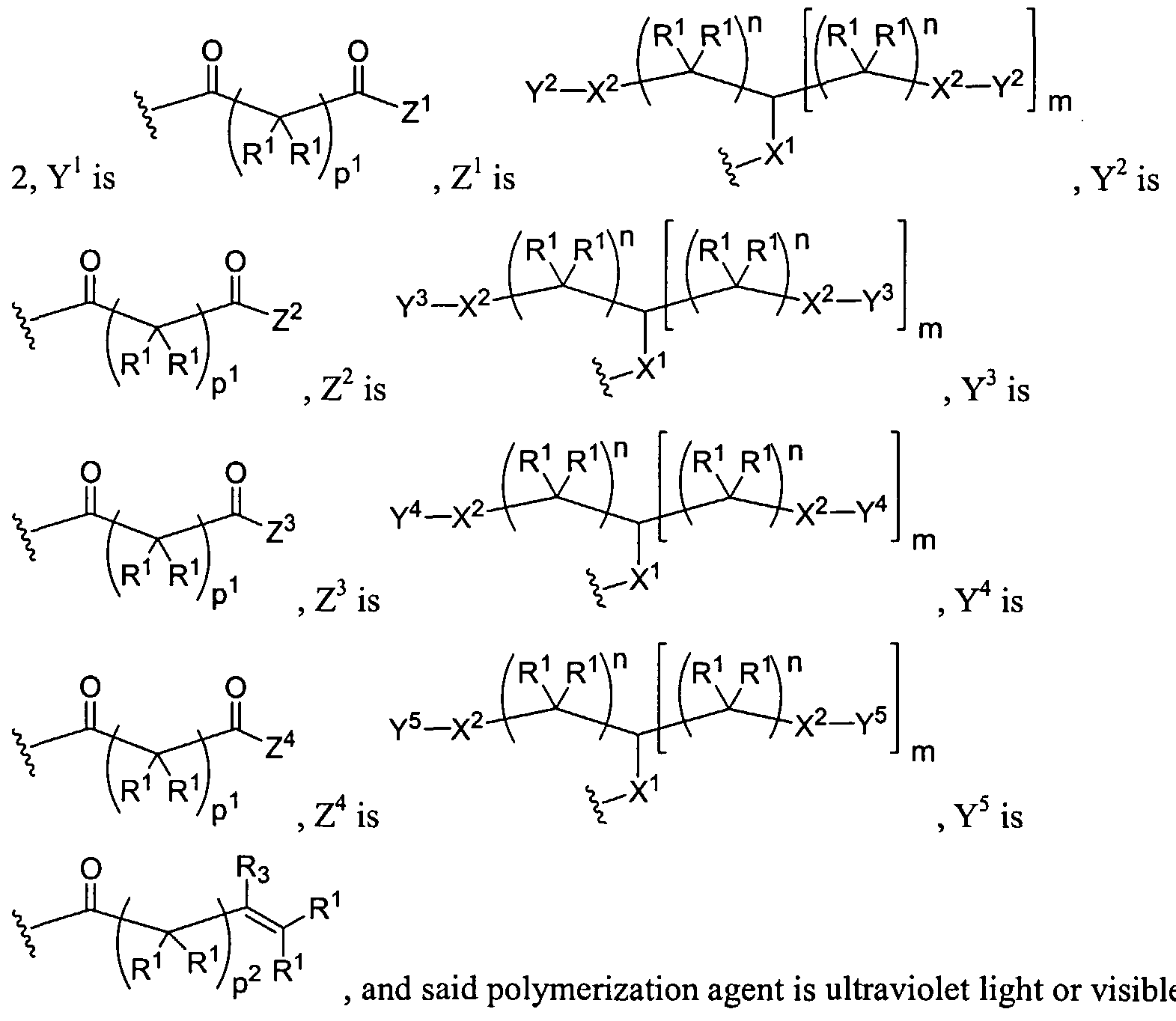
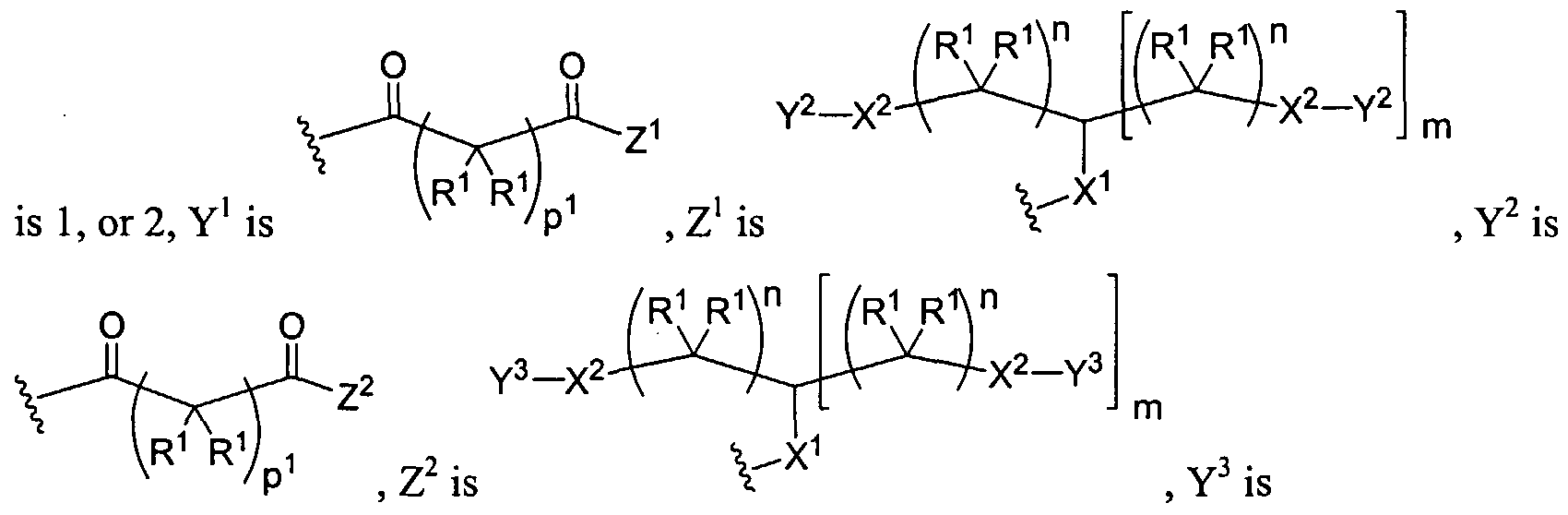
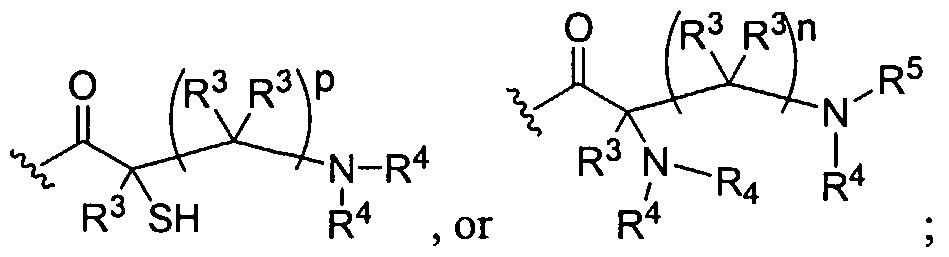
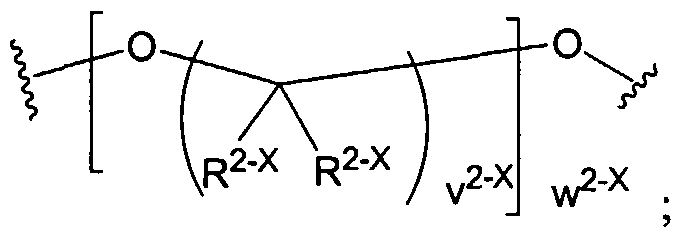
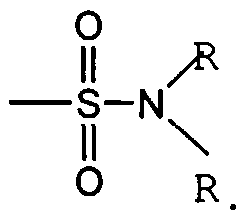
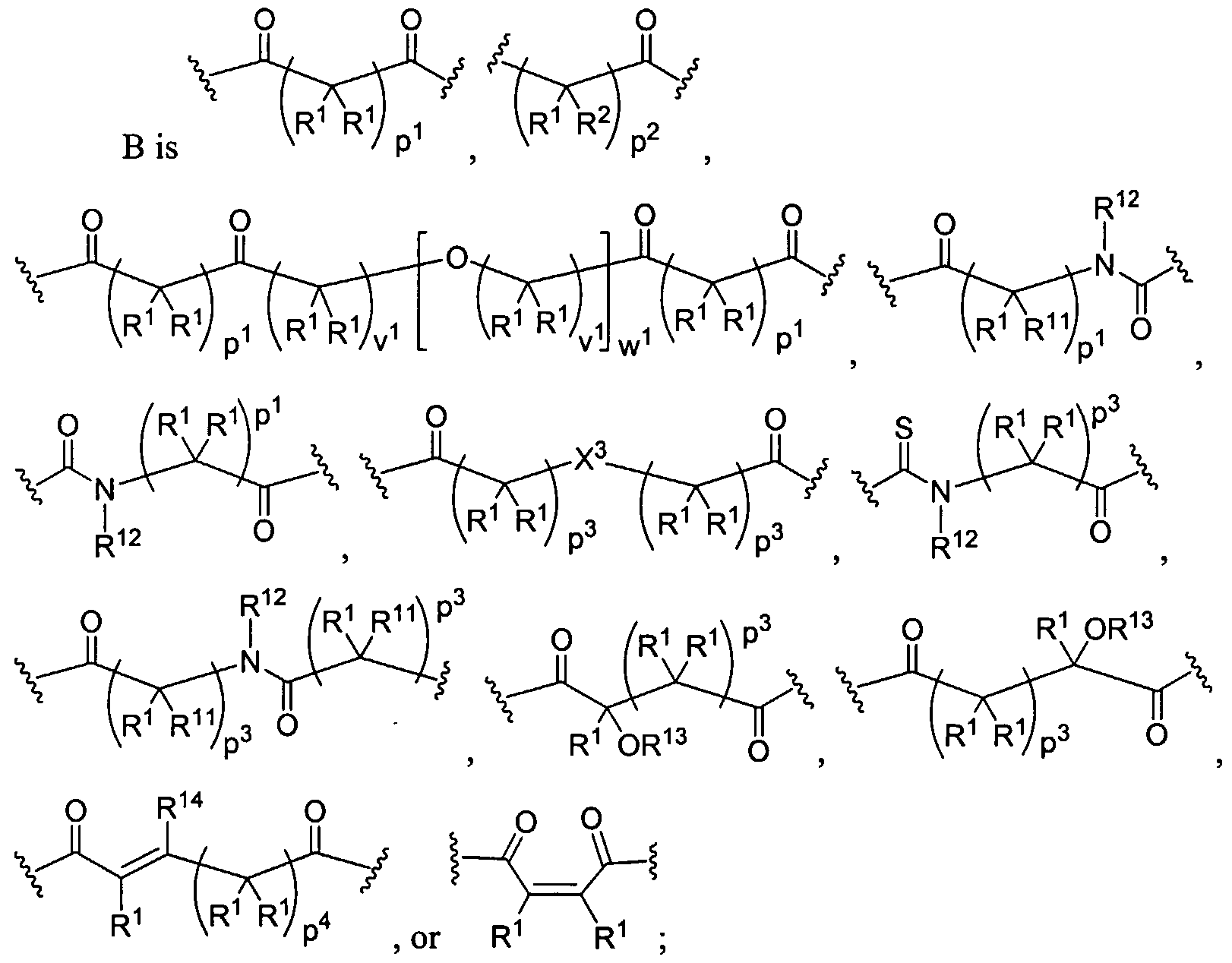
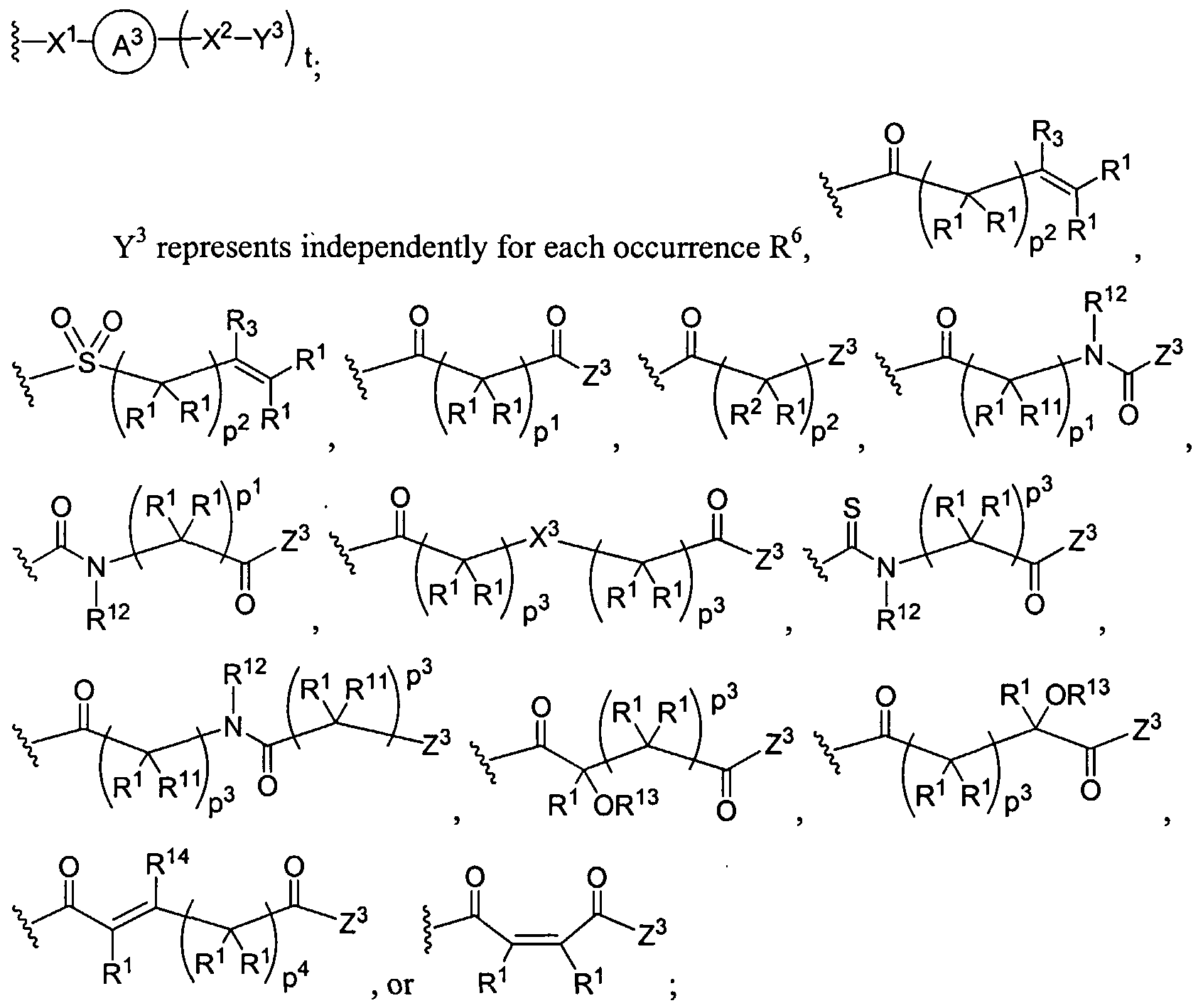
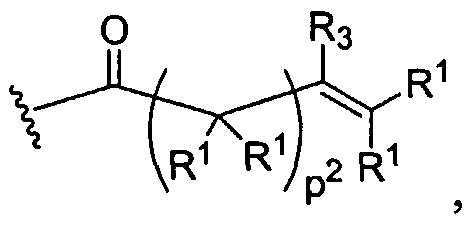
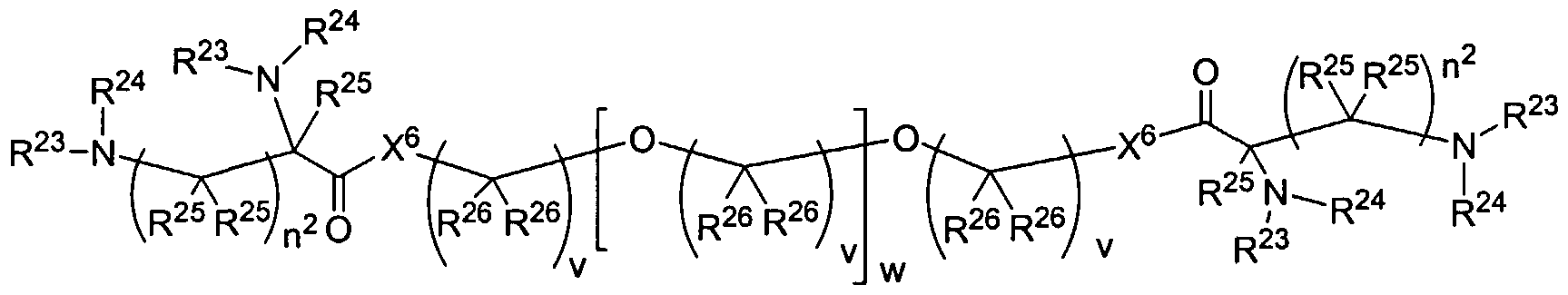
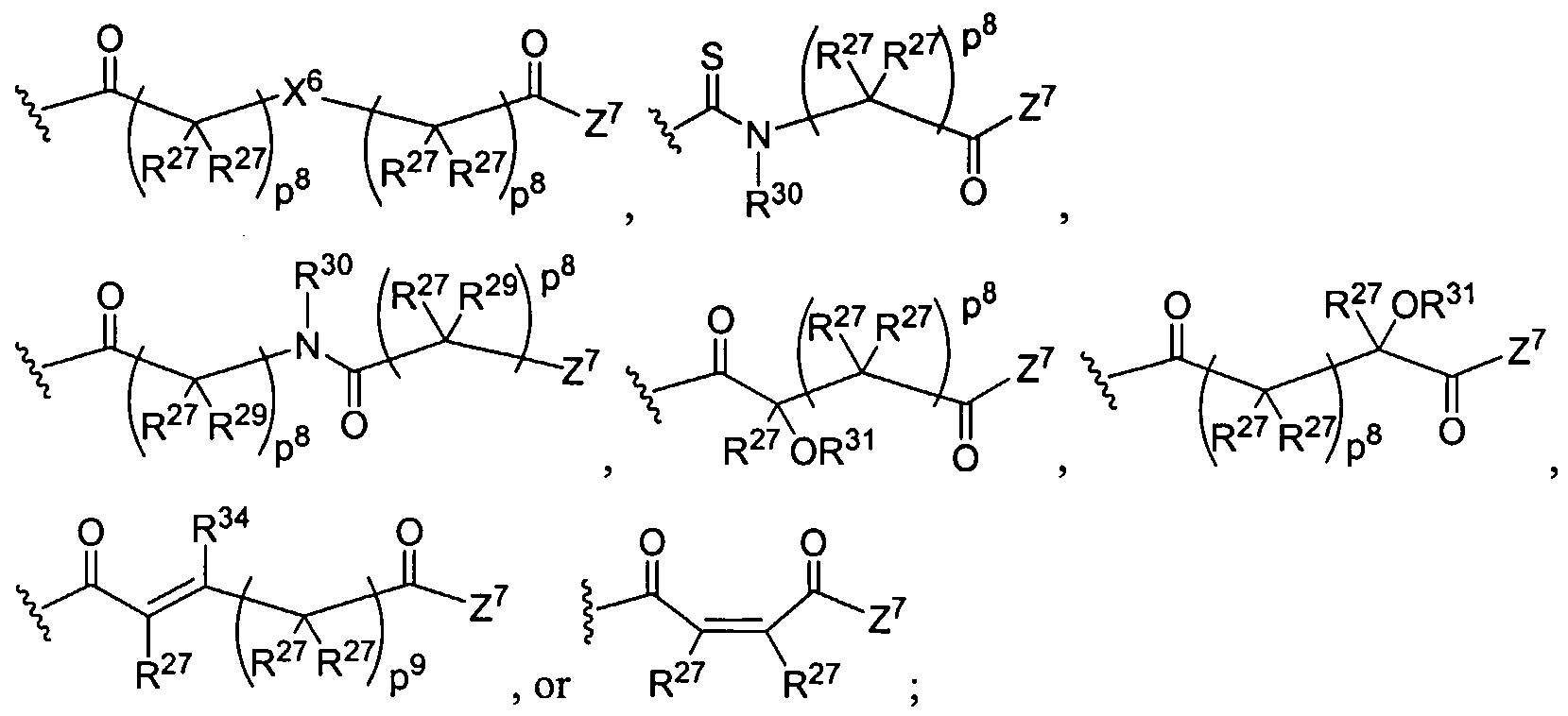
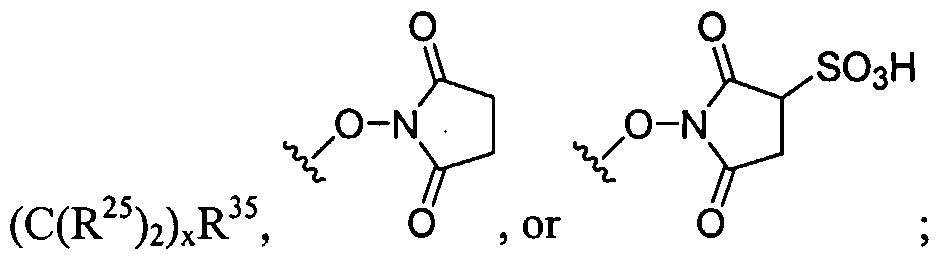
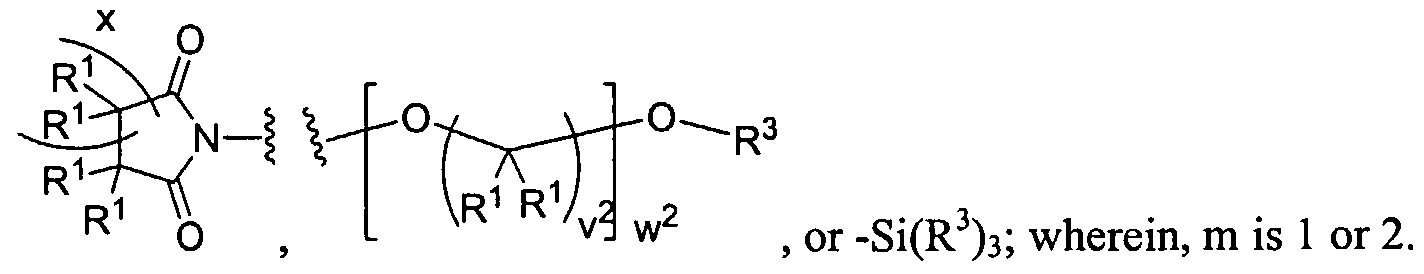
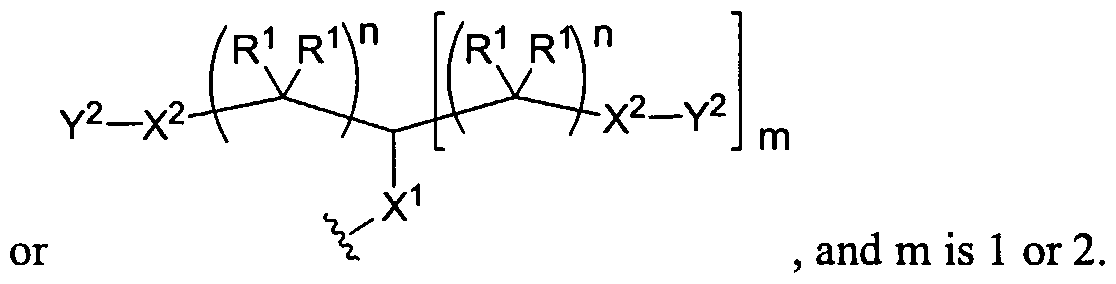
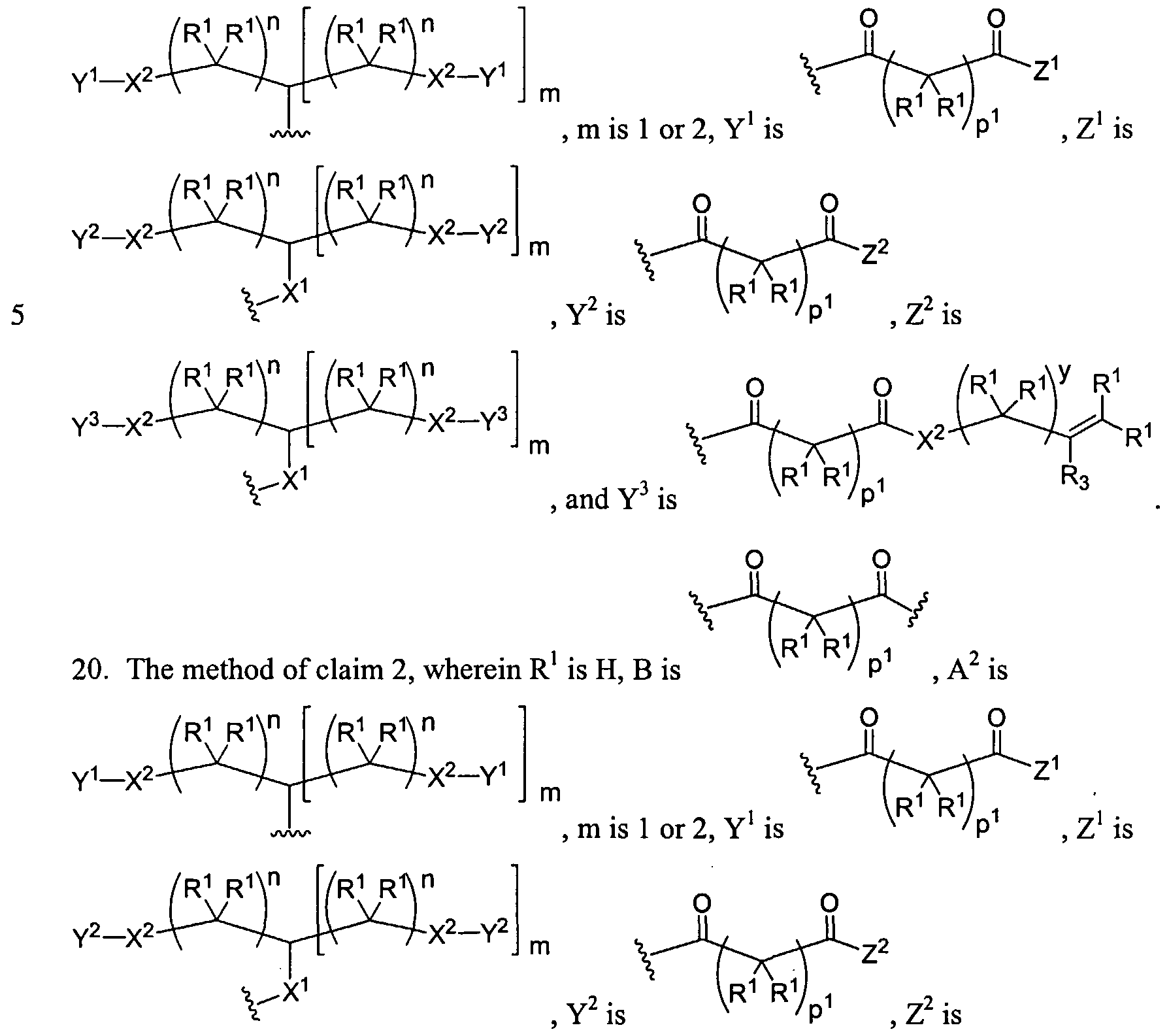
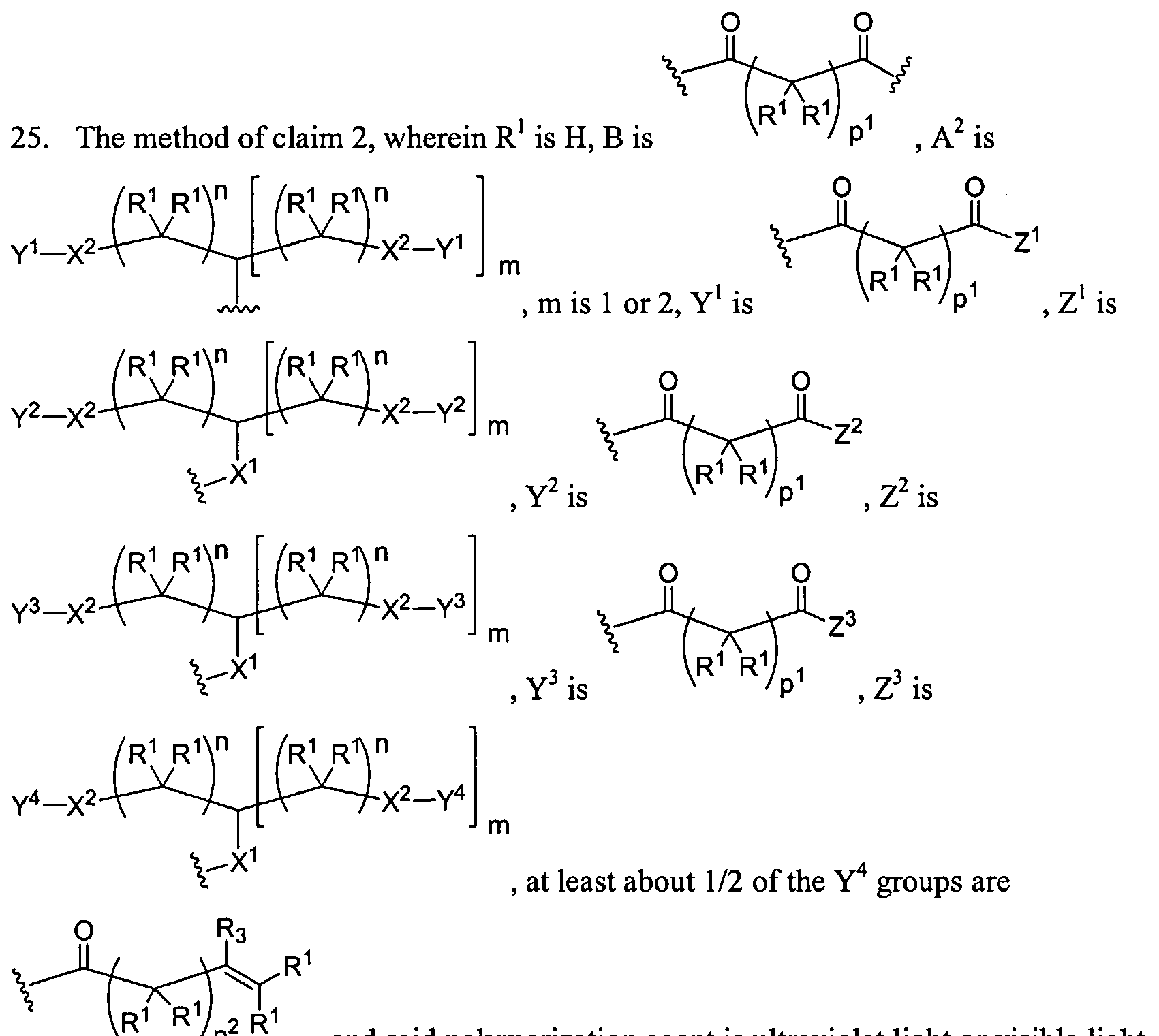
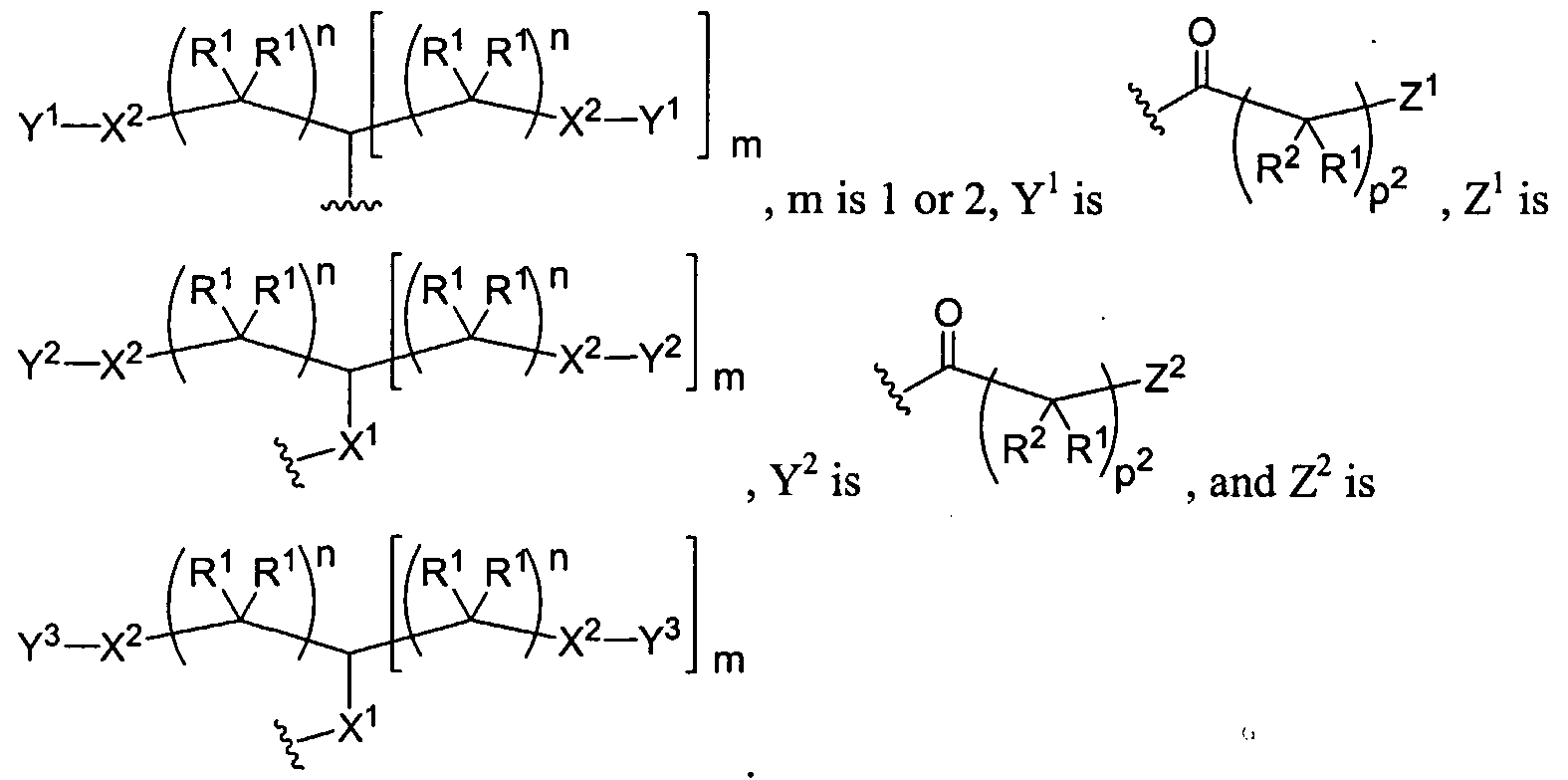
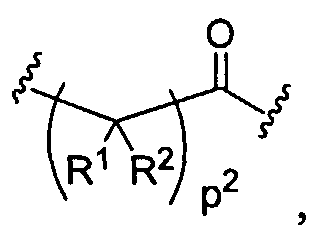
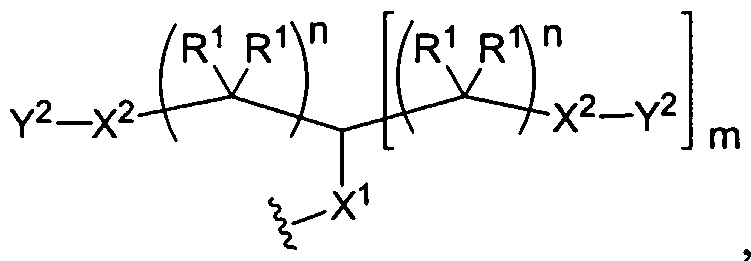
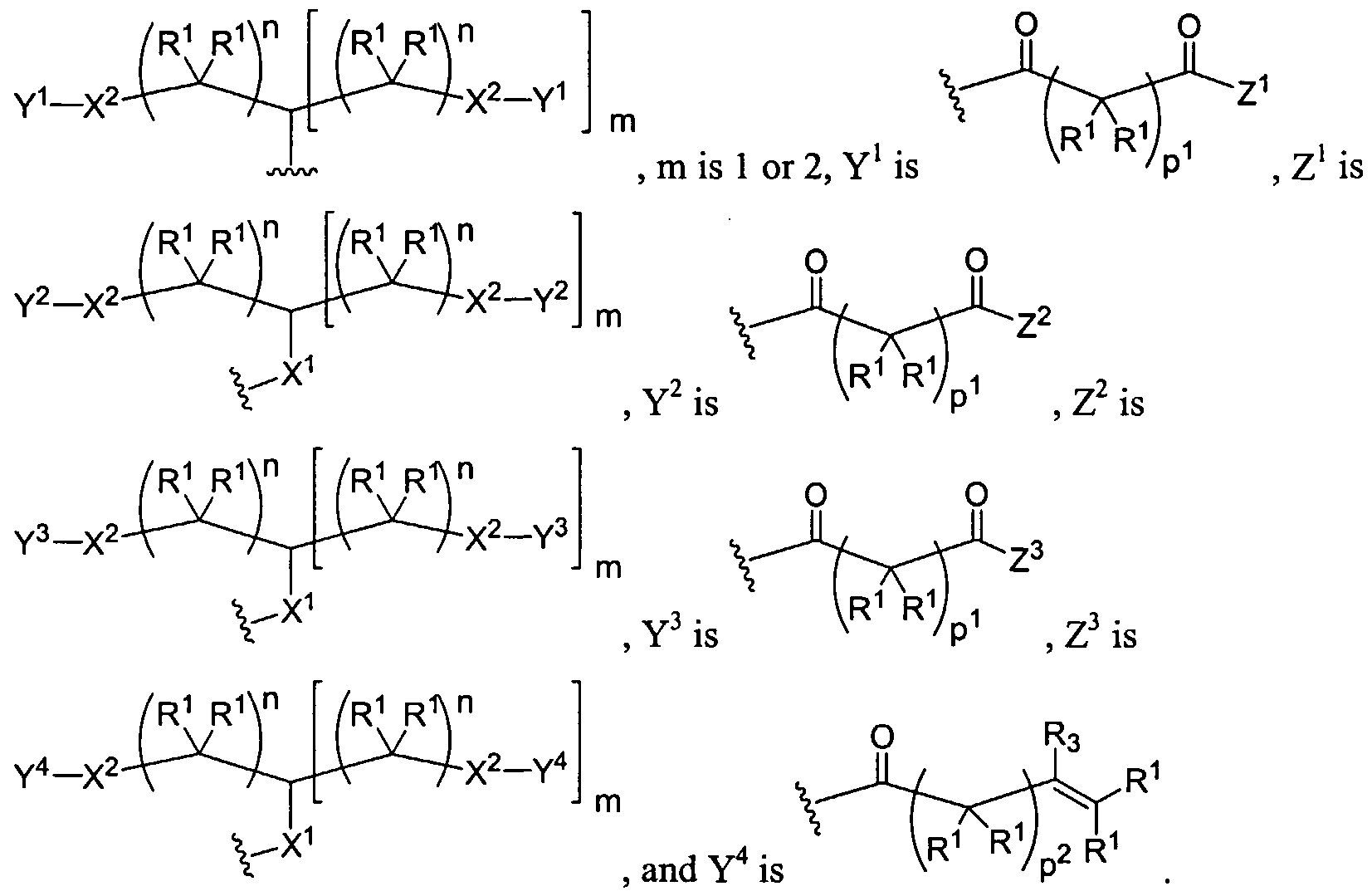
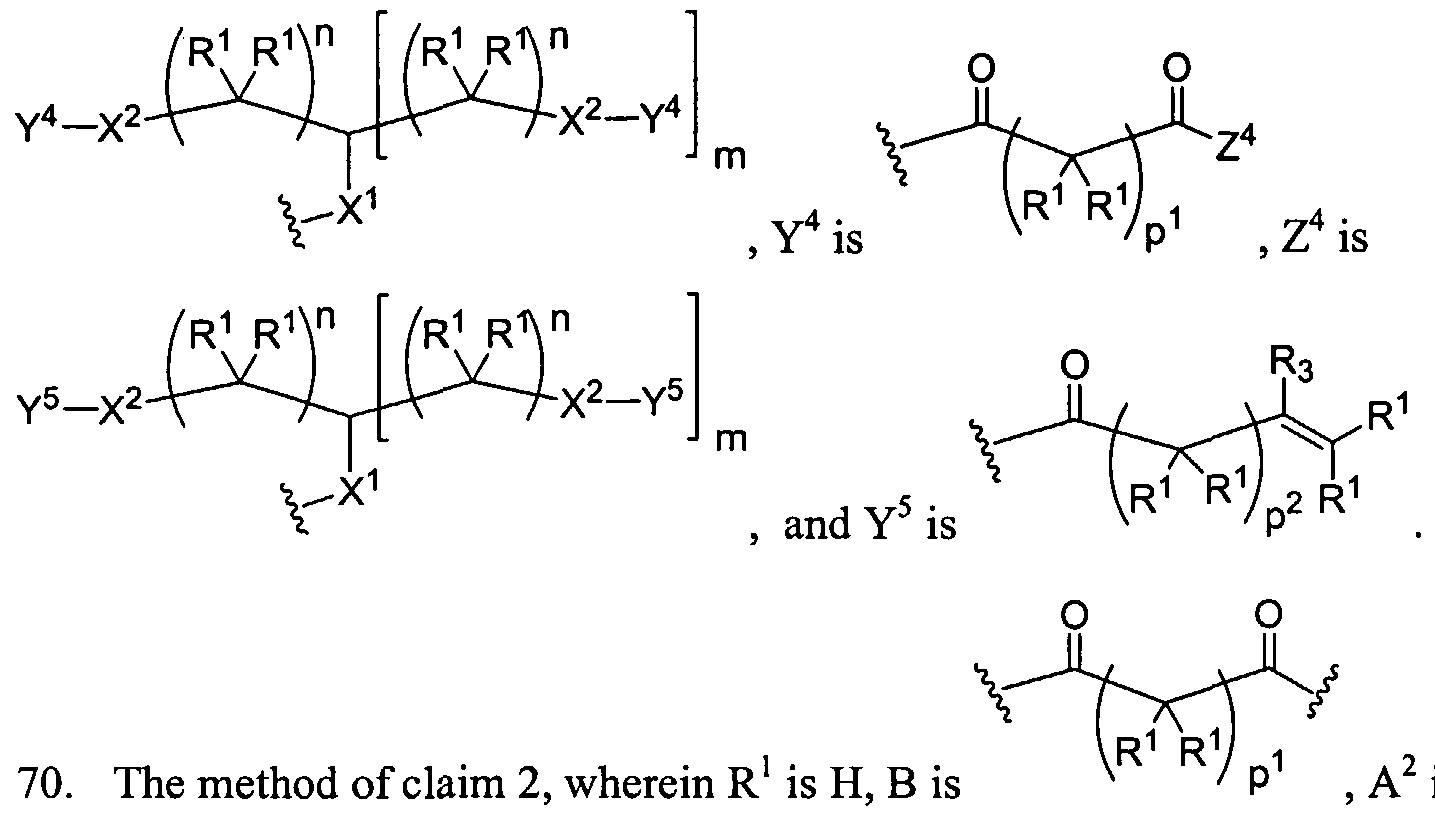
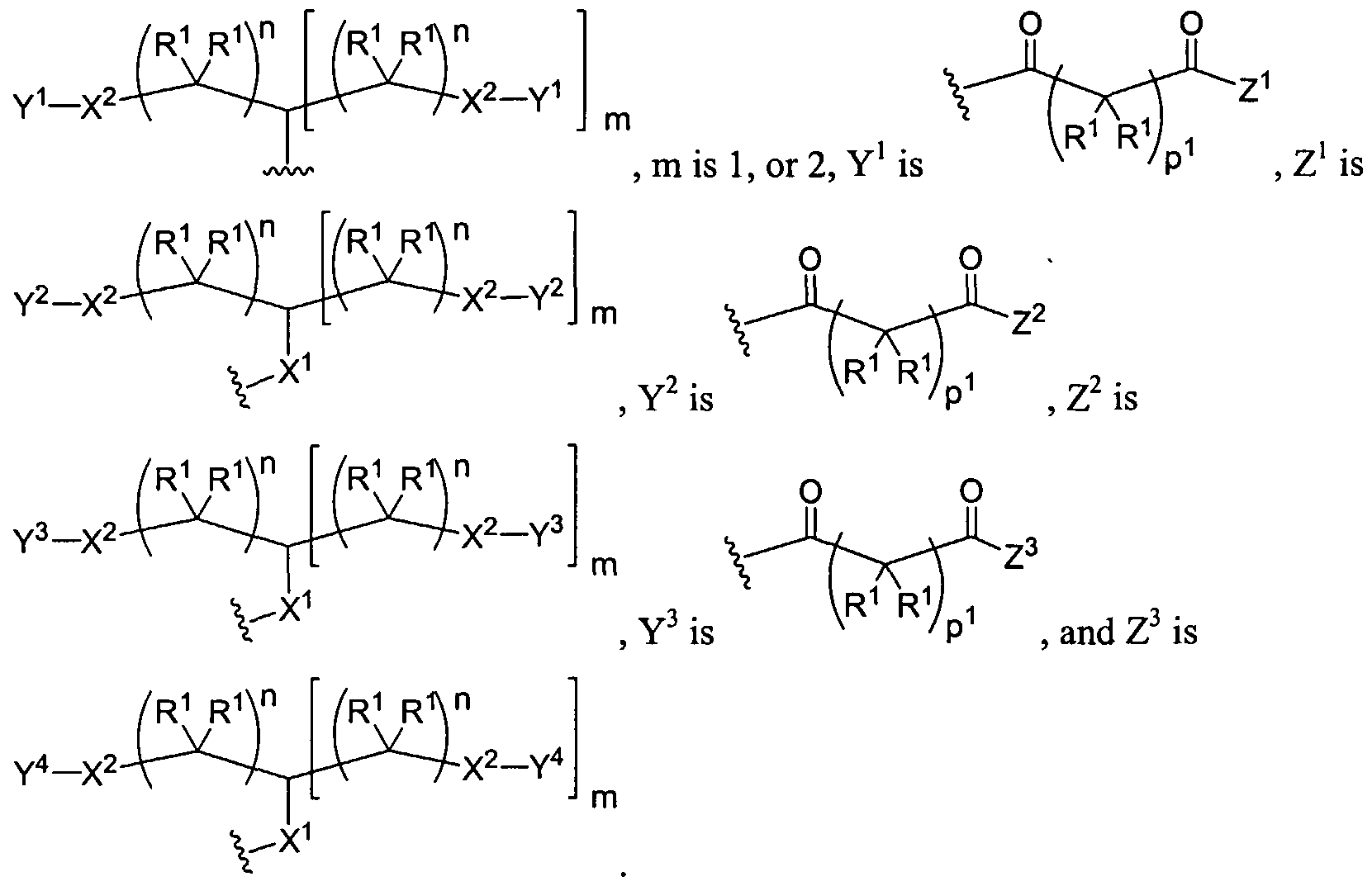
Figure 1 depicts various monomers that can be used to prepare dendrimers used in the invention.
Figure 2 depicts various monomers that can be used to prepare dendrimers used in the invention.
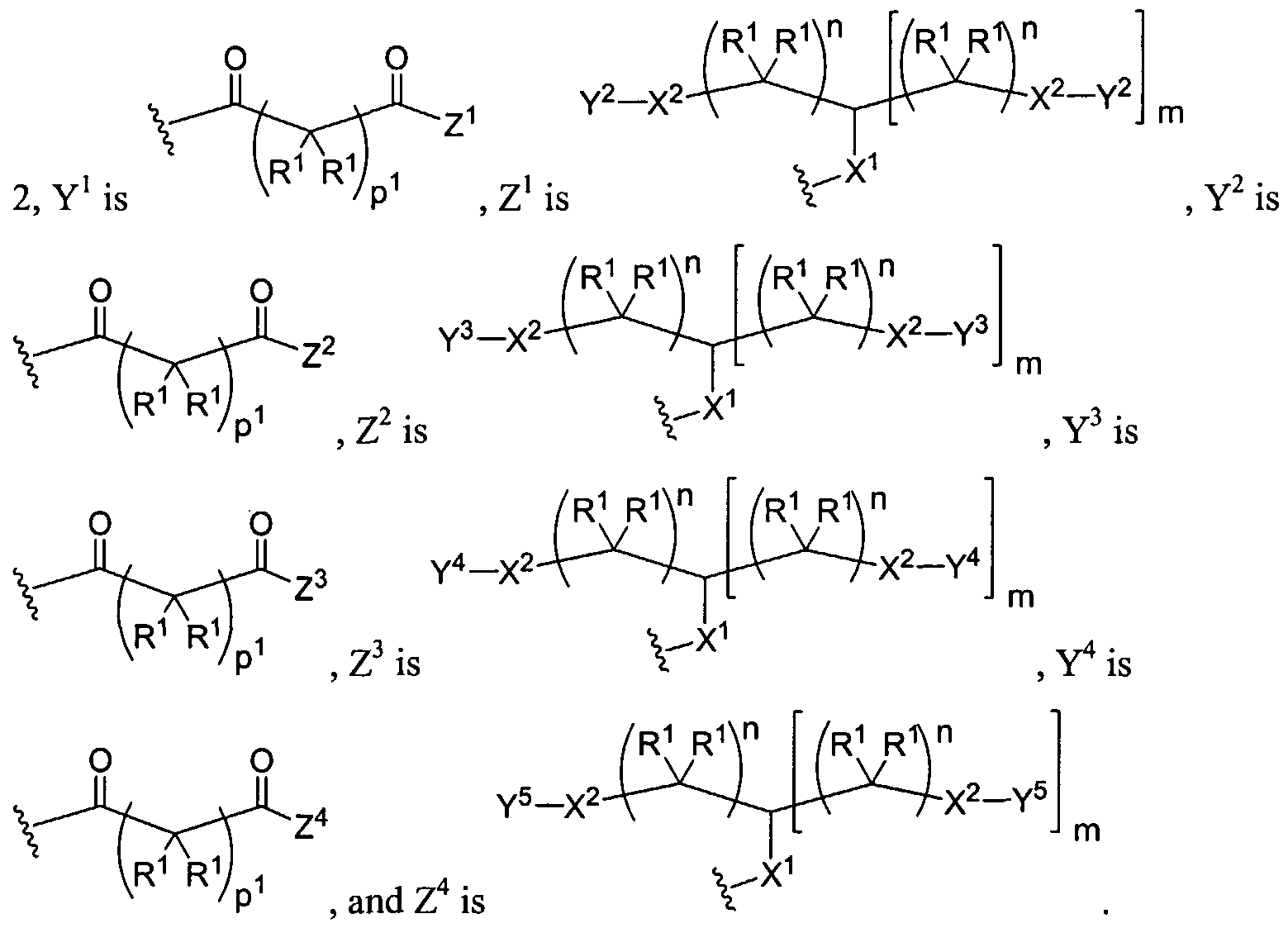
Figure 3 depicts various monomers that can be used to prepare dendrimers used in the invention.
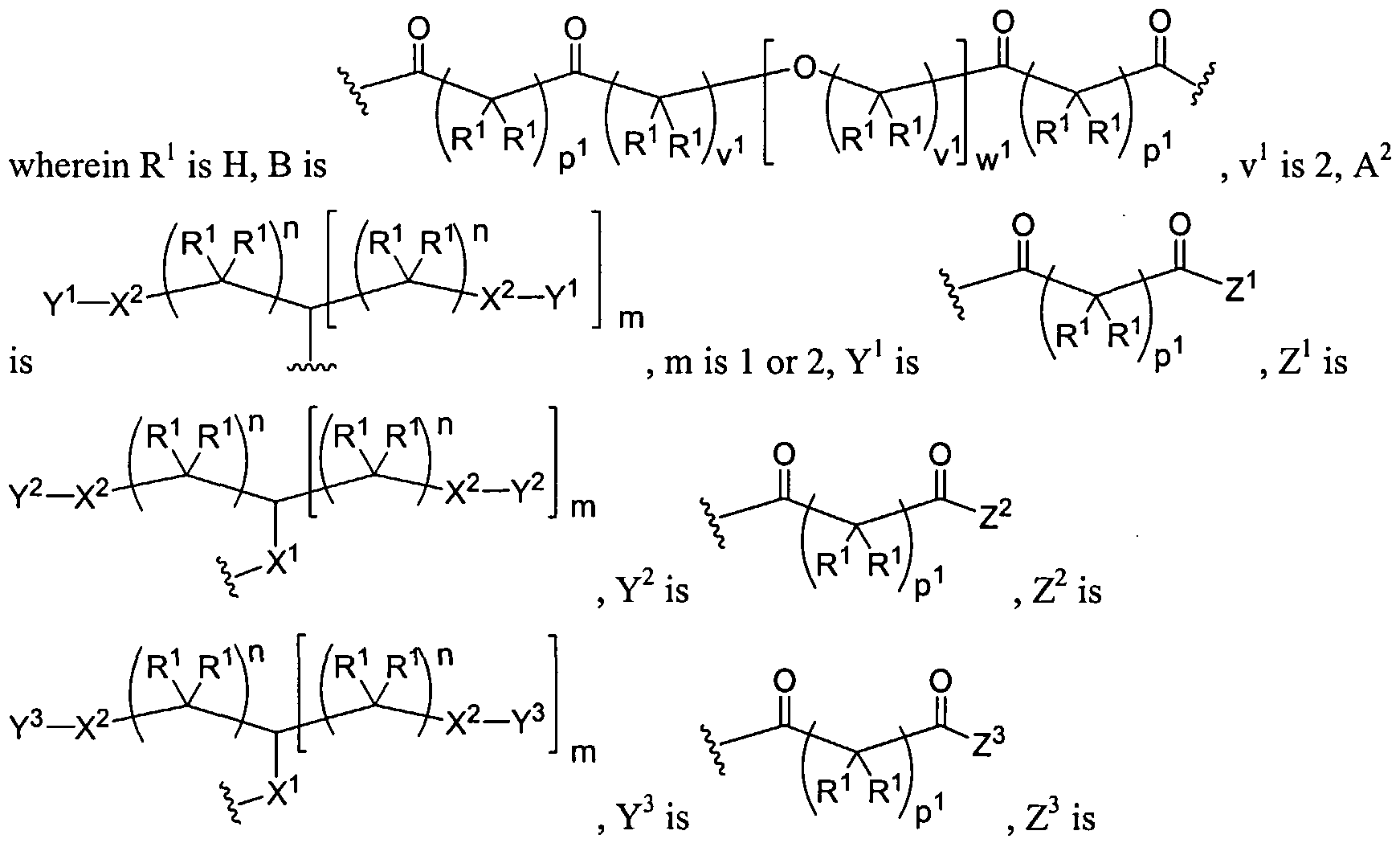
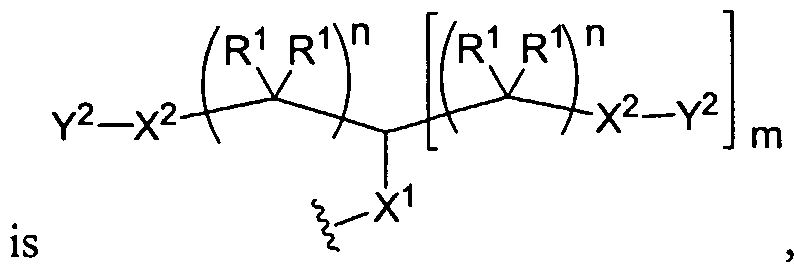
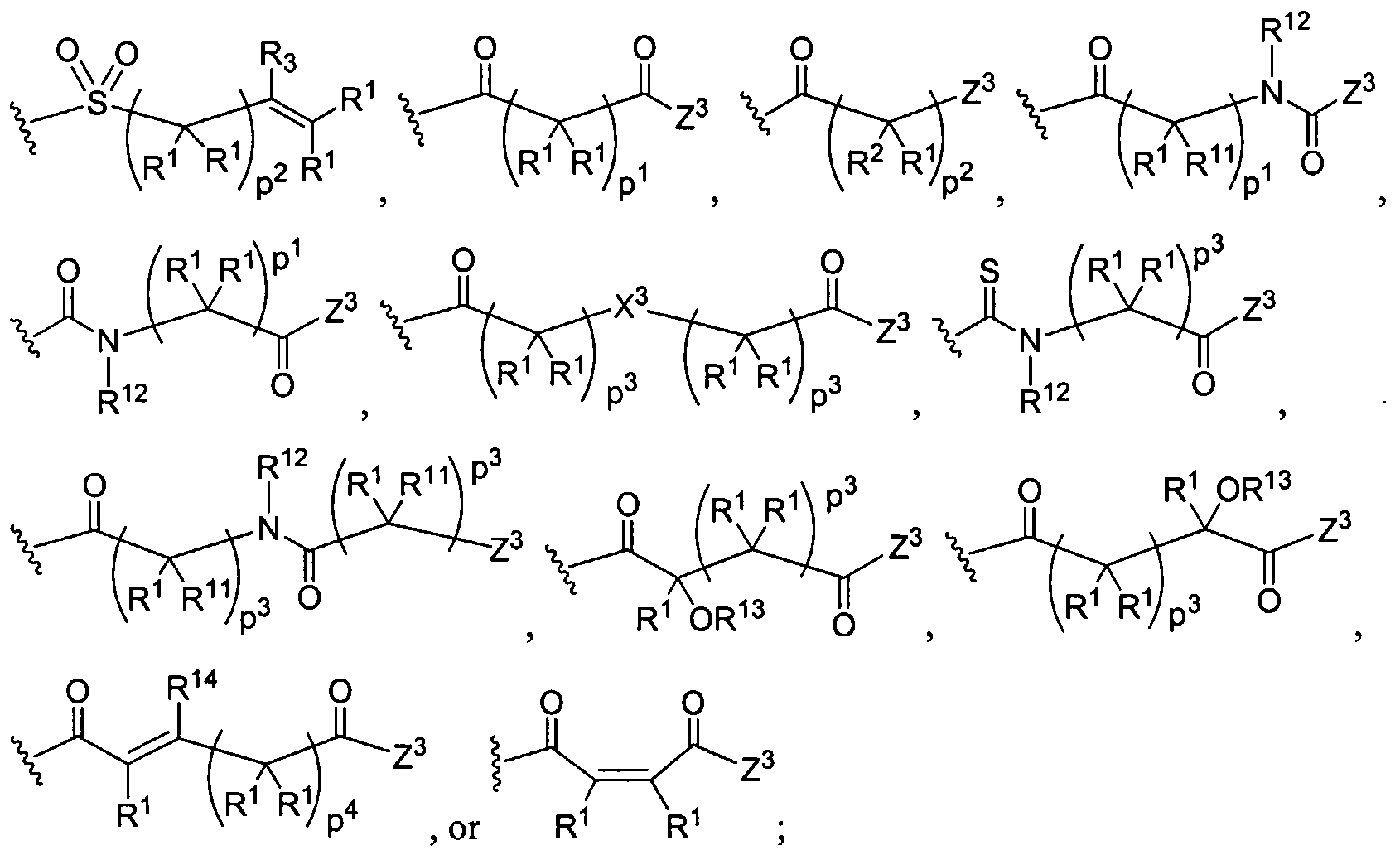
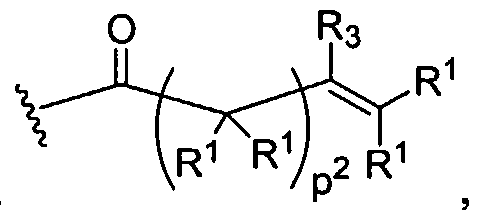
Figure 4 depicts various monomers that can be used to prepare dendrimers used in the invention. Figure 5 depicts various monomers that can be used to prepare dendrimers used in the invention.
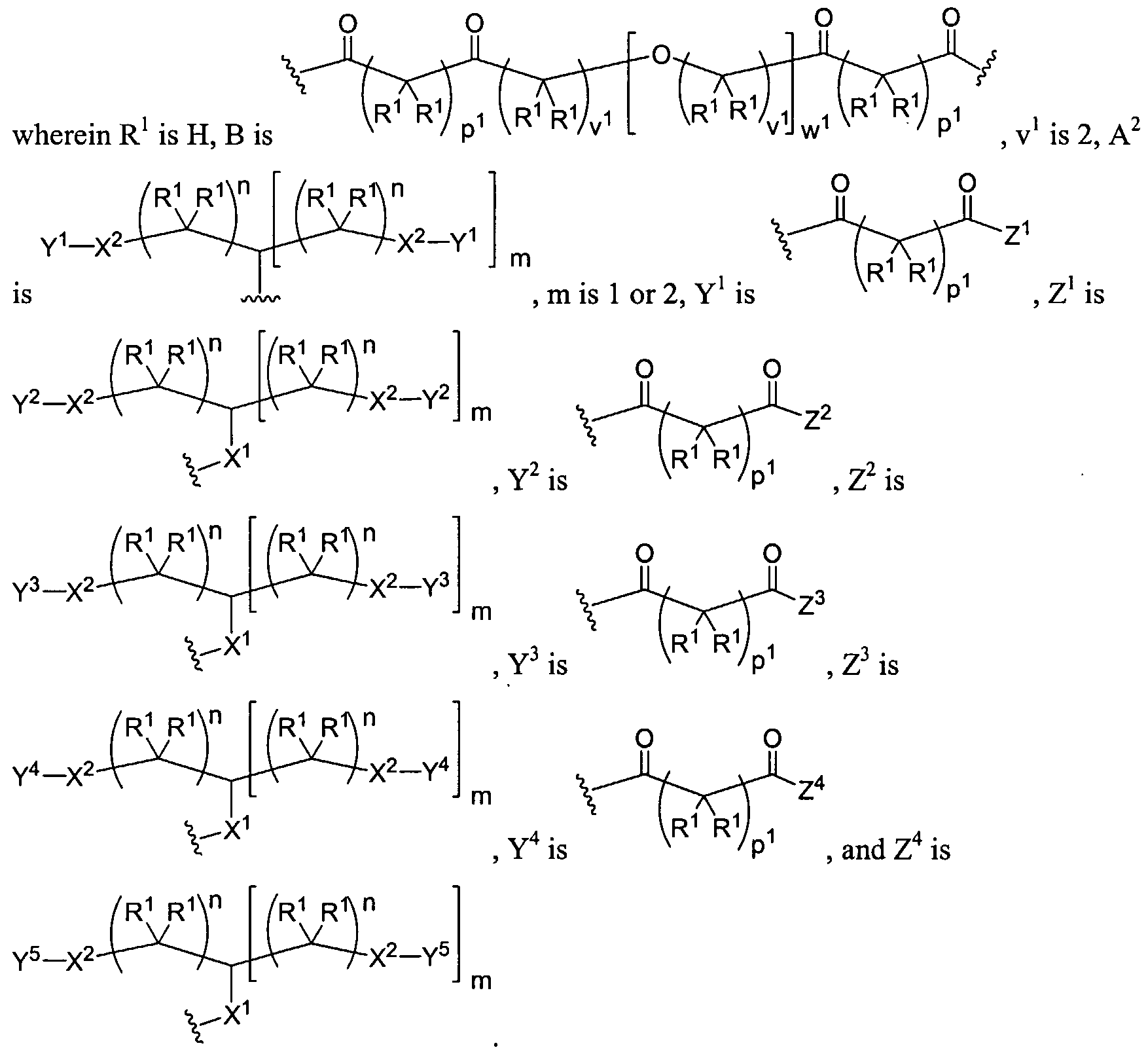
Figure 6 depicts various monomers that can be used to prepare dendrimers used in the invention.
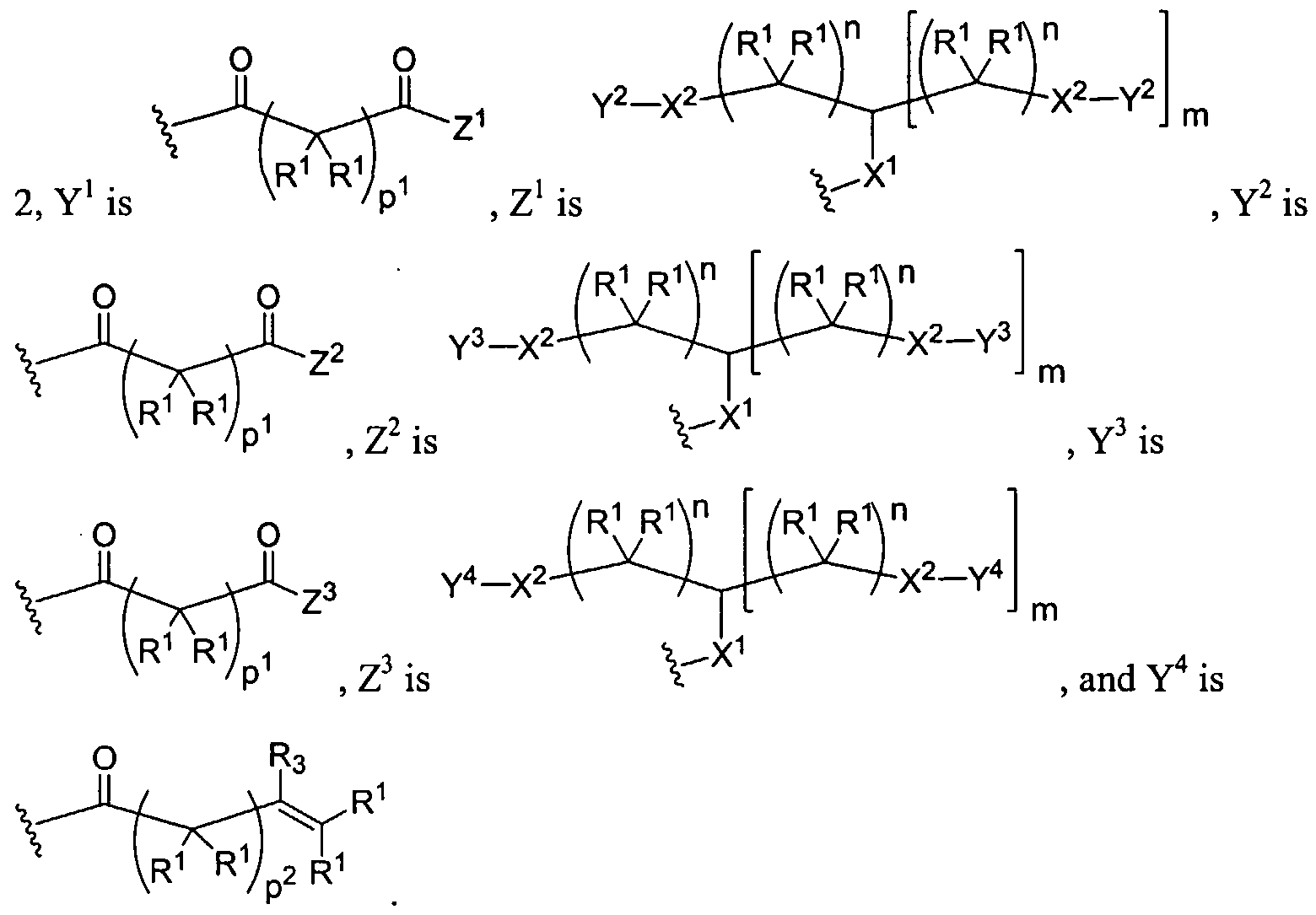
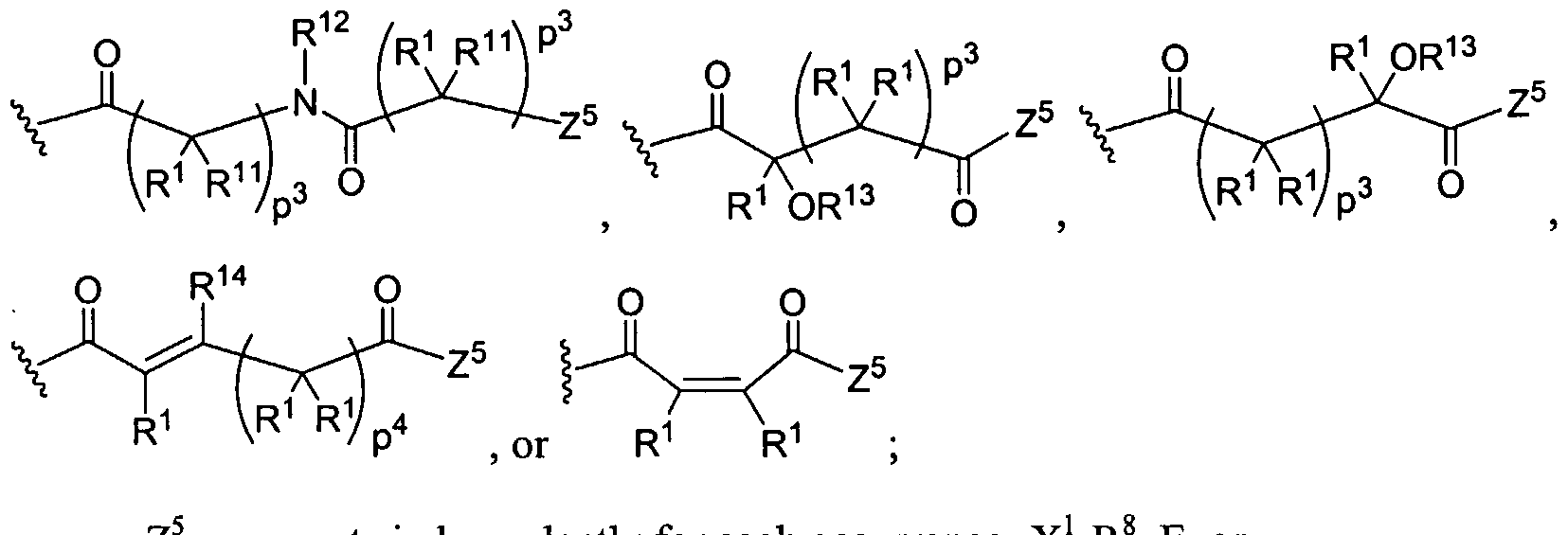
Figure 7 depicts various monomers that can be used to prepare dendrimers used in the invention.
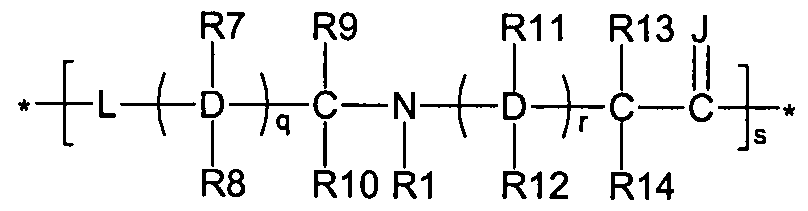
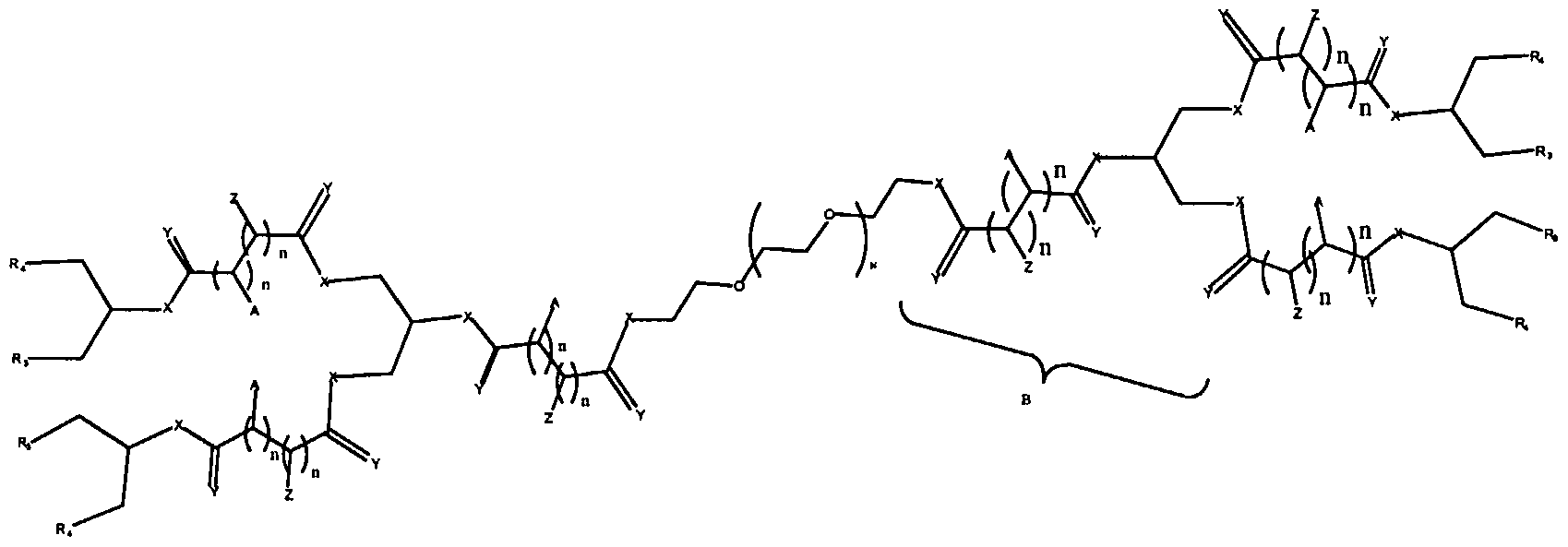
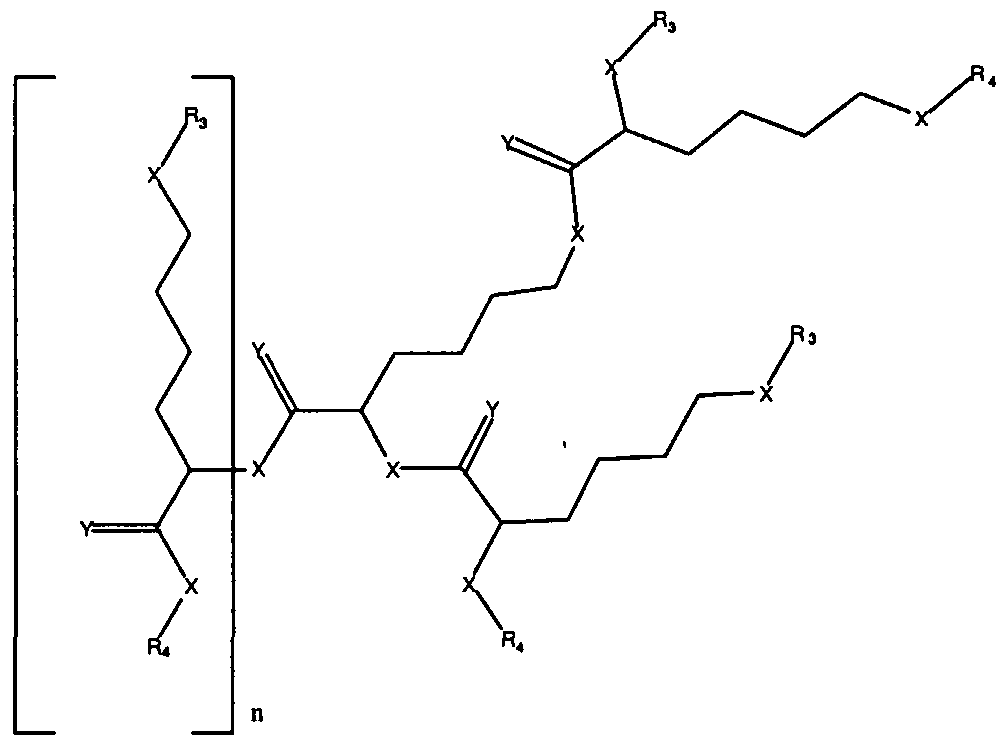
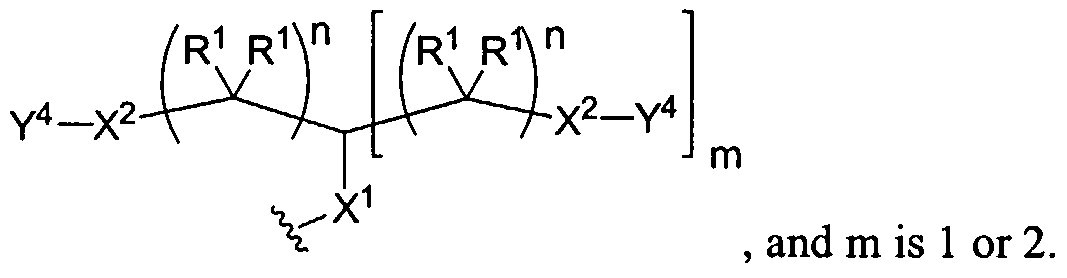
Figure 8 depicts a dendrimer terminated with nucleoside groups amenable to the invention.
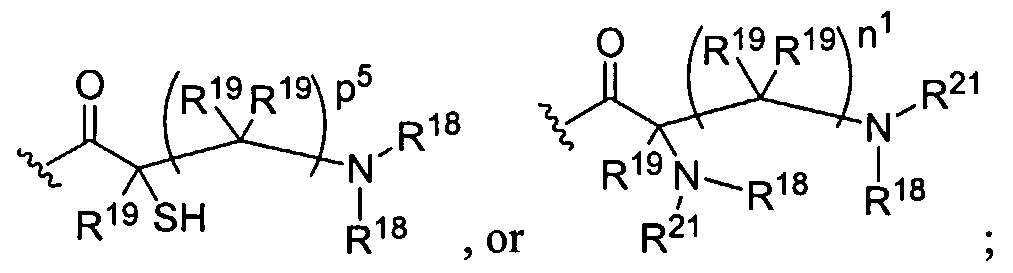
Figure 9 depicts dendrimers and compounds useful for making dendrimers amenable to the present invention. Figure 10 depicts a dendrimer amenable to the present invention.
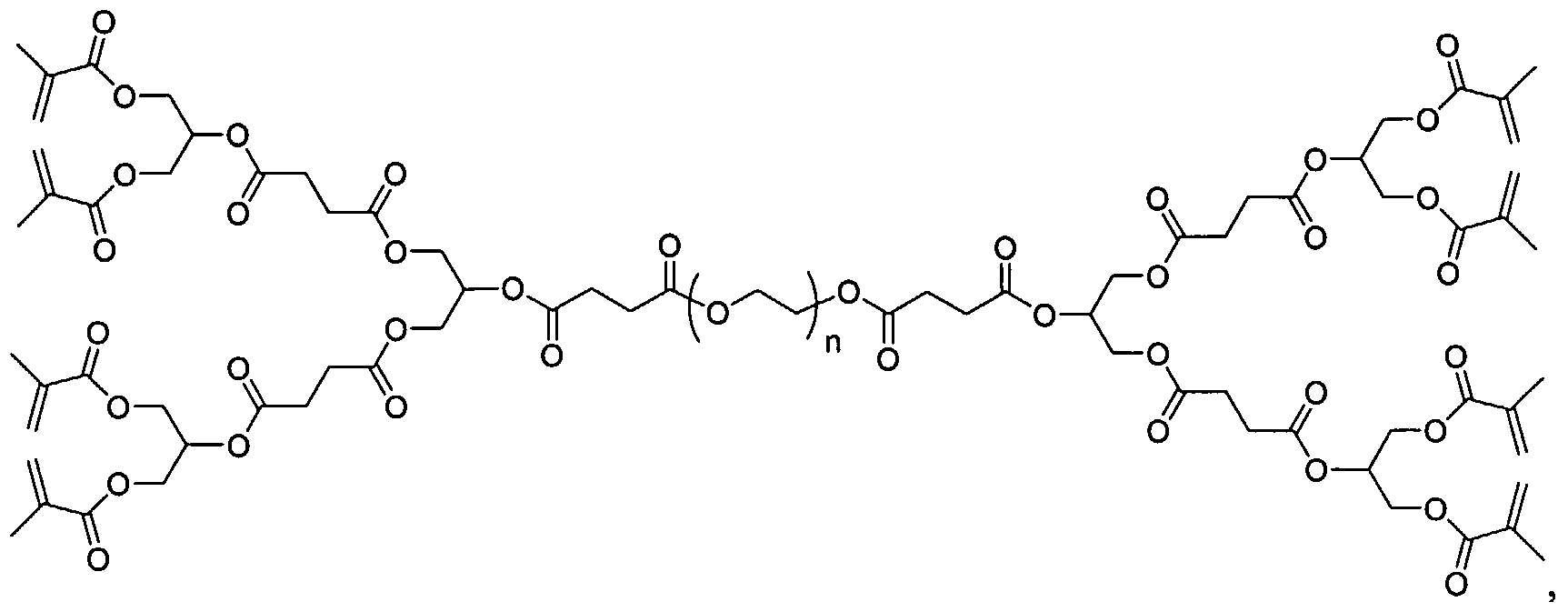
Figure 11 depicts photocrosslinkable PEG34oo-(PGLSA-MA4)2 macromer 1 for hydrogel formation.
Figure 12 depicts the normalized weight of hydrogel samples at 7.5, 10, and 15 % macromer concentration (n = 3), stored in PBS (left, PBS = Dulbecco's Phosphate Buffer Saline) or chondrocyte culture medium (right), at 37 0C, as a function of time.
Figure 13 depicts the complex modulus \G*\, storage modulus G', loss modulus G" and loss angle J of crosslinked hydrogel samples at 4 different concentrations of macromer 1 in PBS.
Figure 14 depicts a determination of the compressive modulus E, as a function of concentration of macromer 1 in PBS. The compression-relaxation experiment for 20 % 1 is shown on the left, the linear curve fits for all concentrations and the resulting compressive modulus E is shown on the right.
Figure 15 depicts histological sections of hydrogels with 7.5 % or 15 % macromer 1 after 2 or 4 weeks incubation. Top: Proteoglycans were stained in the Safranin-0 sections; collagen was stained in the Masson's Trichrome sections. Bottom: Type I and type II collagen were immunostained; no significant type I collagen was detected at either concentration. The length of the inserted bar is 100 μm.
Detailed Description of the Invention The present invention generally relates to the use of dendritic polymers in treating defects in cartilaginous tissue. The dendrimers are treated with a polymerization agent to form a polymeric gel. The dendrimer-based hydrogels provide a temporary scaffold for new tissue growth, can be used to deliver growth factors, can be used with cartilage or bone cells, and can be used with stem cells to regenerate tissue. The dendritic polymers of the invention provide a lower viscosity solution, a higher mechanical-strength network, a lower weight-percent polymer requirement, and a greater water content when crosslinked than end-functionalized linear polymers. The properties of the dendrimers of the invention provide significant advantages over existing polymers for in situ forming scaffolds, as well as the ability to confer diverse functionalization of the dendritic branches. In certain instances, the dendritic macromolecules of the invention are used in conjuction with a natural polymer such as HA, collagen, or GAG fragments such that a hydrogel is formed that contains both the dendritic components and the natural polymer. The dendritic polymers/macromolecule compositions of the invention are useful in orthopedic surgery. Specifically, the dendritic polymers/macromolecule compositions of the invention can be used to repair articular cartilage. Importantly, repair of cartilaginous tissue is just one
orthopedic application, and one skilled in the art can readily determine the utility of these polymers and their hydrogels for other orthopedic applications.
The dendritic compounds of the invention contain a reactive functional group on the terminus of the dendrimer that undergoes reaction with a polymerization agent to form a repair agent used to treat the cartilage defect. In certain instances, the reactive functional group is an acrylate group or a nucleophile. Dendrimeric compounds functionalized with acrylate groups polymerize under the influence of ultraviolet light. In certain instances, a photoinitiator is added to the monomelic dendrimer. A large number of photoinitiators are known in the art and are amenable to the present invention. Representative photoinitiators include ethyl eosin, eosin Y, fluorescein, 2,2-dimethoxy-2 -phenyl acetophenone, 2- methoxy-2-phenylacetophenone, camphorquinone, rose bengal, methylene blue, erythrosin, phloxime, thionine, riboflavin, methylene green, acridine orange, xanthine dye, and thioxanthine dyes. In a preferred embodiment, the photoinitiator is eosin Y. The polymerized dendrimeric composition can be used to repair a cartilage defect. In certain instances, the reactive functional group on the terminus of the dendrimer is a cysteine group. The cysteine functionalized dendrimers form polymers when treated with a compound containing multiple electrophilic groups, such as an aldehyde or an activated ester. In certain instances, the compound bearing the electrophilic groups is a polyethylene glycol. A large number of chemical compounds can be used to prepare the core and branching portions of the dendrimer. For example, the core and/or branching portion of the dendrimer may be derived from succinic acid, adipic acid, glycolic acid, or lactic acid. In certain instances, the dendritic macromolecules possess two or more different linkages within the macromolecule. For example, the dendrimer may be composed of glycerol, succinic acid, and/or glycine residues and the aforementioned residue is bonded to an adjacent group to form an ester or carbamate. In other instances, the dendritic macromolecule is a dendritic-linear hybrid wherein the linear core is a polyethylene glycol and the dendritic wedges contain ester and/or carbamate linkages. Another aspect of the invention relates to dendritic macromolecules comprising hydrogen bonding linkages within the dendrite framework. The hydrogen bonding linkages, e.g., amide or carbamate,
enable non-covalent interations between the dendritic polymer and proteins, glycoproteins, and the like.
The polymers, after being crosslinked, can be seeded with cells and then used to repair the damaged cartilaginous tissue. Alternatively, the polymers and cells can be mixed and then injected into the in vivo site and crosslinked in situ for tissue repair or replacement. The crosslinked polymers provide a three dimensional template for new cell growth. Crosslinking, such as with a methacrylated functionalized denditic polymer, can be achieved using light or a chemical reaction. An embodiment of this invention is the preparation of crosslinkable biodendritic macromolecules that can undergo a covalent or non-covalent crosslinking reaction to form a three-deminsional crosslinked gel or network, wherein the crosslinking reaction does not involve a single or multi-photon process (i.e., light). The dendritic polymer can be used for the encapsulation of or the covalent attachment of pharmaceutical agents/drugs such as bioactive peptides (e.g., growth factors), antibacterial compositions, antimicrobial compositions, and anti-inflammatory compounds to aid/enhance repair of the cartilaginous tissue.
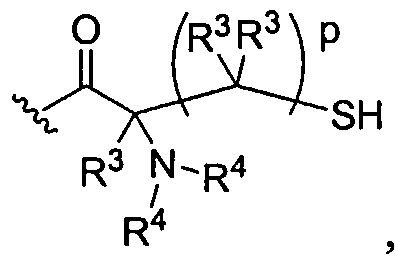
The cartilage defect can be filled using the photocrosslinkable dendritic macromolecule and subsequently photocrosslinked with light to afford the hydrogel. Alternatively, the defect is filled with a self-gelling dendritic system. For example, a Iys3cys4 dendron can be mixed with a polyethylene glycol containing two or more reactive electrophilic groups, e.g., NHS-activated ester, malemide, or aldehyde, where a gel is formed quickly, e.g., within 1 minute.
Hydrogel materials are particularly successful as tissue engineering scaffolds because they are water-saturated turgid networks that mimic the three-dimensional environment of cells in native cartilaginous tissues. In addition, the high water-content allows for rapid diffusion of nutrients and oxygen to, and waste products and carbon dioxide from, the cells. This rapid diffusion has a positive influence on the metabolic activity of cells within the scaffold material.
The requirements for a successful tissue-engineering scaffold for cartilage repair are complex and extend beyond basic biocompatibility and low toxicity. As a stress-absorbing tissue, the mechanical properties of cartilage define its function in the body, and are thus an important selection criterion for a cartilage repair material. Cartilaginous tissues, including
articular cartilage, meniscus and intervertebral disk, are porous, water saturated viscoelastic materials that exhibit high mechanical stiffness. The mechanical properties reflected in the compressive and (dynamic) shear moduli strongly depend on the specific tissue-type. See Table 1 and the following references: Setton, L. A.; Elliot, D. M.; Mow, V. C, Osteoarthr. Cartilage 1999, 7, 2-14; Setton, L. A.; Mow, V. C; Howell, D. S., J. Orthop. Res. 1995, 1(4), 437-82; Iatridis, J. C; Weidenbaum, M.; Setton, L. A.; Mow, V. C, Spine 1996, 21(10), 1174-1184; and Umehara, S.; Tadano, S.; Abumi, K.; Katagiri, K.; Ukai, T., Spine 1996, 21(7), 811-819.
Table 1. Mechanical properties of a selection of native cartilaginous tissues in humans.
Compressive Modulus Complex Shear Modulus Phase
£/ kPa |G*| / kPa Angle δl °
Articular Cartilage 600 440, 600-1000 13
Meniscus n/a 100 22
Annulus Fibrosus 111, 76 540 n/a
Nucleus Pulposus 5.8 7-21 23-31
Restoring mechanical function at the time of cartilage repair is highly desirable for maintaining tissue and joint function, reducing inflammation at the trauma site, and maintaining the environment governing cell metabolism and matrix homeostasis. See Guilak, F.; Kapur, R.; Sefton, M. V.; Vandenburgh, H. H.; Koretsky, A. P.; Kriete, A.; O'Keefe, R. J., Ann. K Y. Acad. ScL 2002, 961, 207-209. Consequently, there is significant interest in manipulating the mechanical properties of a biomaterial during synthesis and implantation in order to obtain a functional scaffold that will support load and permit integration with native tissue. In addition to mechanical function, several other properties may affect scaffold success, including diffusion of large and small molecules, porosity, surface properties, tissue adhesion, morphology, and (bio)degradation kinetics.
Photocrosslinkable hydrogels based on poly(ethylene glycol) dimethacrylate have shown considerable promise as a tissue-engineering scaffold. See Elisseeff, J.; Mclntosh, W.; Anseth, K.; Riley, S.; Ragan, P.; Langer, R., J. Biomed. Mater. Res. 2000, 57 (2), 164- 171 ; Elisseeff, J.; Anseth, K.; Sims, D.; Mclntosh, W.; Randolph, M.; Langer, R., Proc. Natl. Acad. ScL U. S. A. 1999, 96 (6), 3104-3107; and Elisseeff, J.; Anseth, K.; Sims, D.; Mclntosh, W.; Randolph, M.; Yaremchuk, M.; Langer, R., Plast. Reconstruct. Surg. 1999, 104 (4), 1014-1022. The in situ photocrosslinking ability of these systems is highly desirable in a cartilage tissue-engineering application for a variety of reasons. First, it allows the uncrosslinked macromer solution to be mixed with cells or soluble factors, such as growth factors or cytokines, prior to delivery to the defect site. Second, the uncrosslinked macromer solution can easily flow into irregularly shaped defects common to damaged or diseased cartilage, facilitating integration with the surrounding native tissue. Third, the liquid state of the macromer solution allows access to surgically inaccessible trauma sites via endoscope-assisted (micro)surgery. Lastly, these materials, once crosslinked in situ, provide immediate adhesion and mechanical integrity to the defect site at the time of implantation.
A significant limitation of the poly(ethylene glycol) dimethacrylate hydrogels, however, is the lack of (bio)degradation, which hampers long-term viability of incorporated cells and inhibits formation of neocartilage throughout the scaffold. One solution to this problem is the introduction of degradation sites into these hydrogels, allowing chondrocytes to degrade the scaffold while extracellular matrix is deposited. It was shown recently by Hubbell and Anseth that both linear oligopeptides and linear esters allow biodegradation when incorporated into the scaffold design. See Halstenberg, S.; Panitch, A.; Rizzi, S.; Hall, H.; Hubbell, J. A., Biomacromolecules 2002, 3 (4), 710-723; Bryant, S. J.; Anseth, K. S., J. Biomed. Mater. Res. 2003, 64A (1), 70-79; Martens, P. J.; Bryant, S. J.; Anseth, K. S., Biomacromolecules 2003, 4 (2), 283-292; and Bryant, S. J.; Durand, K. L.; Anseth, K. S., J. Biomed. Mater. Res. 2003, 67 A (4), 1430-1436. However, it is crucial that degradation of the matrix material is tuned to the synthesis of extracellular matrix in such a way that the mechanical properties of the site are not compromised. See Wilson, C. G.; Bonassar, L. J.; Kohles, S. S., Arch. Biochem. Biophys. 2002, 408 (2), 246-254.
The role of the temporary scaffold is multifaceted in cartilage repair, and since current clinical procedures are limited, new and alternative designs, methods, and materials are highly sought after. Dendrimers are highly branched, well-defined macromolecules that are ideal compounds for the assembly of such materials. See Frechet, J. M. J. Proc. Natl. Acad. ScL U. S. A. 2002, 99 (8), 4782-4787; Frechet, J. M. J.; Tomalia, D. A. Dendrimers and other dendritic polymers . John Wiley & Sons: New York, 2002; p 648; Grayson, S. M.; Frechet, J. M. J. Chem. Rev. 2001, 101 (12), 3819-3867; Newkome, G. R.; Moorefield, C. N.; Vogtle, F. Dendrimers and dendrons: concepts, synthesis, perspectives. Wiley-VCH: Weinheim, 2001; Bosman, A. W.; Janssen, H. M.; Meijer, E. W. Chem. Rev. 1999, 99 (7), 1665-1688; Fisher, M.; Vogtle, F. Angew. Chem. Int. Ed. 1999, 38 (7), 884-905; Majoral, J. P.; Caminade, A. M. Chem. Rev. 1999, 99 (3), 845-880; Matthews, O. A.; Shipway, A. N.; Stoddart, J. F. Prog. Polym. ScL 1998, 23 (1), 1-56; Zheng, F.; Zimmerman, S. C. Chem. Rev. 1997, 97 (5), 1681-1712; Newkome, G. R.; Moorefield, C. N.; Keith, J. M.; Baker, G. R.; Escamilla, G. H. Angew. Chem. Int. Ed. 1994, 33 (6), 666-668; Issberner, J.; Moors, R.; Vogtle, F., Angew. Chem. Int. Ed. 1994, 33, 2413-2420; Vogtle, F. Dendrimers. Springer: Berlin, 1998; Vol. 197, p 240; Vogtle, F. Dendrimers II: Architecture, Nanostructure and Supramolecular Chemistry. Springer: Berlin, 2000; Vol. 210, p 311; and Vogtle, F., Dendrimers III: Design, Dimension, Function. Springer: Berlin, 2001; Vol. 212, p 198. Dendritic polymers provide a multivalent and modular base for the design and optimization of novel macromers for tissue engineering scaffold applications. The branched structure allows considerable degradation before the crosslinked network breaks down, maintaining mechanical integrity during degradation. Branched structures, through their multivalency, also allow higher crosslink densities at low concentrations compared to linear functionalized polymers, providing the potential to achieve the seemingly conflicting requirements of high mechanical strength and high water content critical for cartilage repair. In addition, the well-defined nature of these macromolecules allows analysis of structure-property relationships between the molecular features of the macromers and the mechanical and physiochemical properties of the hydrogel constructs, allowing subsequent structure-based optimization of these constructs for specific applications.
Biocompatible dendrimers, or føodendrimers, constructed from moieties known to be biocompatible have been reported. See Grinstaff, M. W. Chem. Eur. J. 2002, 8 (13), 2839-2846. Tissue adhesives based on methacrylated block copolymers, consisting of a polyethylene glycol (PEG) core and biodendrimer wedges synthesized from glycerol and succinic acid, display excellent corneal tissue adhesion and show considerable promise as an ocular sealant for sutureless eye-surgery. See Carnahan, M. A.; Grinstaff, M. W., J. Am. Chem. Soc. 2001, 123 (12), 2905-2906. These compounds can be used as in situ photocrosslinkable scaffold materials for articular cartilage tissue engineering. See Figure 11. The physical characterization of the biodendrimer scaffolds including swelling, mechanical and degradation properties, as well as the ability of the hydrogel to support articular chondrocytes and extracellular matrix synthesis in vitro are reported herein.
The results from the preparation and analysis of the physical properties of various hydrogels are described below. Concentrations of macromer 1 (Figure 11) from 5 % w/w to 20 % w/w were observed to crosslink uniformly within the molds. The hydrogel samples formed from biodendrimer solutions below 7.5 % w/w were fragile after crosslinking, however, and were consequently difficult to investigate. Thus, we limited our study to macromer concentrations from 7.5 to 20 %. The appearance of the crosslinked hydrogel pellets was dependent on macromer concentration. Equilibrated hydrogel constructs formed from lower concentrations of the macromer (7.5 and 10 %) were slightly opaque, whereas the hydrogel pellets formed from higher macromer concentrations were transparent (15 and 20 %). This likely reflects differences in the ratio of intra- and intermolecular crosslinking between the lower and higher macromer concentrations, and may indicate some precipitation during polymerization at the lower macromer concentrations.
No appreciable swelling was observed in any of the four concentrations during equilibration at 37 0C in PBS or chondrocyte medium after crosslinking. An initial decrease in normalized sample weight was observed, with re-swelling to 85-100% of the initial sample weight within 7 days (Fig. 2). This is in sharp contrast to linear poly(ethylene glycol) dimethacrylates that can show swelling in excess of 200 %. The hydrogel samples showed no appreciable changes in sample weight while immersed in PBS or chondrocyte medium, up to 35 days, indicating a lack of degradation without the presence of cells
(Figure 12).
The dynamic mechanical properties (G', G", |G*|, and δ) showed no appreciable frequency dependence from 0.1 rad/s up to 100 rad/s, at all four macromer concentrations. The complex modulus \G*\ of the hydrogels showed only limited concentration dependence, increasing from 1.2 to 1.7 kPa, over the concentration range. The loss angle δ, however, increased strongly from 6.3 ± 0.5° at the lowest macromer concentration to 70.6 ± 0.3° at the highest macromer concentration, showing a transition from primarily elastic behavior at the lowest macromer concentration to more viscoelastic behavior at higher macromer concentrations. See Figure 13.
The equilibrium compressive modulus E was determined from the equilibrium compressive stress following a stepwise increase in compressive strain (Figure 14). The compressive modulus increased substantially from 3.7 ± 0.1 kPa at the lowest macromer concentration to 34 ± 5 kPa at the highest macromer concentration, showing strong non¬ linear stiffening of the hydrogels with increasing macromer concentration.
Histological sections prepared from cell-hydrogel constructs showed chondrocytes displaying a rounded morphology in hydrogels prepared from both the 15 and 7.5 % macromer concentrations, throughout the duration of culture. After two weeks of culture,
Safranin-0 and Masson's Trichrome staining indicated that chondrocytes encapsulated in hydrogels at the lower macromer concentration accumulated abundant extracellular matrix rich in proteoglycans and collagen, respectively (Figure 15). In contrast, cells encapsulated in hydrogels at the higher macromer concentration produced extracellular matrix only in the immediate vicinity of each cell. Sections of cell-hydrogel constructs prepared from the lower macromer concentration stained strongly for type II collagen after two weeks suggesting the accumulation of extracellular matrix with molecular components present as found in native articular cartilage. No significant straining for type I collagen was observed (Figure 15). The cell-hydrogel constructs at lower macromer concentration also started showing histological signs of degradation, with evidence of small voids in the hydrogel scaffold. Similar voids were not apparent in the cell-hydrogel constructs formed from the higher macromer concentrations.
After four weeks of culture, the difference between the lower and higher concentration hydrogels was even more striking. The accumulation of proteoglycans and
(type II) collagen in the lower macromer concentration samples had increased significantly and spread throughout the scaffold. The cell-hydrogel constructs formed at lower macromer concentration had lost physical integrity, with some samples disintegrating into several smaller fragments. The loss of integrity was also evident from the larger voids visible in the constructs. These cell-hydrogel constructs were fully degraded in 5-6 weeks. This behavior was not observed for the cell-hydrogel constructs formed at the higher macromer concentration, even after twelve weeks of culture (not shown). Few differences were observed between the time-points in the accumulation of extracellular matrix in the cell-hydrogel constructs at higher macromer concentration after two, four, or even twelve weeks (not shown) of culture.
Hence, hydrogels constructed from chemically crosslinked methacrylated macromer 1, at a range of macromer concentrations, support chondrocyte proliferation and cartilaginous tissue growth in vitro. These hydrogels additionally exhibit mechanical properties superior to previously published crosslinked alginate and hyaluronan hydrogels, and show mechanical performance comparable to non-degradable PEG-based hydrogel systems previously reported in the literature. See LeRoux, M. A.; Guilak, F.; Setton, L. A., J. Biomed. Mater. Res. 1999, 47 (1), 46-53; Smeds, K. A.; Grinstaff, M. W., J. Biomed. Mater. Res. 2001, 54 (1), 115-121; Bryant, S. J.; Anseth, K. S., J. Biomed. Mater. Res. 2002, 59 (J), 63-72; and Elisseeff, J.; Mclntosh, W.; Anseth, K.; Riley, S.; Ragan, P.; Langer, R., J. Biomed. Mater. Res. 2000, 57 (2), 164-171. The mechanical properties of the materials presented here also compare favorably with the properties of the degradable hydrogel material published recently by Bryant and coworkers. See Bryant, S. J.; Anseth, K. S., J. Biomed. Mater. Res. 2003, 64A (1), 70-79; Martens, P. J.; Bryant, S. J.; Anseth, K. S., Biomacromolecules 2003, 4 (2), 283-292; Bryant, S. J.; Durand, K. L.; and Anseth, K. S., J. Biomed. Mater. Res. 2003, 67 A (4), 1430-1436.
Excessive swelling can be detrimental to in vivo applications because cartilage trauma sites are generally confined in geometry. When a hydrogel scaffold is formed in situ at the cartilage trauma site, the crosslinked material may swell beyond the boundaries of the trauma and may eventually detach from the wound site or exacerbate the trauma. The biodendrimer-based materials presented here demonstrated only small volume changes during crosslinking and subsequent equilibration at 37 0C in PBS or chondrocyte medium.
The materials studied by Bryant et al., based on methacrylated ?LA-block-PEG-block-PLA copolymers, have favorable mechanical properties, but show volumetric swelling of 150— 200 % with increased swelling during the degradation process. See Bryant, S. J.; Anseth, K. S., J. Biomed. Mater. Res. 2003, 64A (1), 70-79; Martens, P. J.; Bryant, S. J.; Anseth, K. S., Biomacromolecules 2003, 4 (2), 283-292; and Bryant, S. J.; Durand, K. L.; Anseth, K. S., J. Biomed. Mater. Res. 2003, 67A (4), 1430-1436. The lack of swelling in the biodendrimer-based hydrogel materials confirms that these materials are superior for application as scaffolds for in situ tissue engineering in confined defect sites. Although not to be bound to any particular theory, the low swelling ratio is likely a consequence of the multivalent nature of the macromer molecule. This feature allows multiple crosslinks per molecule leading to a higher crosslinking density compared to a bifunctional linear molecule.
In addition to the favorable mechanical properties and low swelling, the biodendrimer-based hydrogel scaffolds support cartilaginous extracellular matrix production. Encapsulated chondrocytes show no signs of dedifferentiation; they retain their rounded morphology and produce extracellular matrix similar to native articular cartilage, including type II collagen and proteoglycans. The lower macromer concentration hydrogel scaffolds were especially supportive of cartilaginous extracellular matrix synthesis. The more rapid synthesis of proteoglycans and collagen by chondrocytes encapsulated in the lower macromer concentration hydrogel may be due to beneficial diffusion characteristics for nutrients, waste products, oxygen, and carbon dioxide, in a hydrogel with higher water content. The scaffold degradation rate is another important parameter for a tissue- engineering material. The degradation rate of the hydrogel-cell constructs at 7.5 % macromer concentration is rapid, with rates comparable to the PLA-PEG-PLA hydrogels. The hydrogel-cell constructs at the higher macromer concentration do not show appreciable degradation, even after 12 weeks in culture, impeding cell proliferation and matrix deposition. The differences in degradation kinetics are likely due to crosslink density. The metabolic activity of the encapsulated chondrocytes, secreting hydrolytic enzymes is another important factor. The negligible degradation in the absence of cells illustrates that the presence of cells has a profound influence on degradation kinetics.
The results obtained with the biodendrimer hydrogel support the strategy of using a dendritic macromolecule to control hydrogel properties and promote cartilaginous tissue formation. Even though the mechanical properties of the lower concentration hydrogels are not identical to native articular cartilage, it appears that simply increasing the macromer concentration to improve mechanical properties is likely not the best route to a better performing scaffold. The degradation rate of the biodendrimer-based hydrogel system will need to be tuned to extracellular matrix deposition, to allow in vivo formation of neocartilaginous tissue before the matrix has fully degraded. It is clear that a compromise between targeted mechanical, diffusion properties, and biochemical properties is necessary to afford an optimal tissue engineering scaffold for cartilage matrix production in vitro or in vivo. The photocrosslinked scaffold presented here allows for the variation of generation, degree of branching, crosslink density, hydrolysable linkage, and end-group functionality, allowing tailoring of the physical, (bio)chemical, and mechanical properties of the scaffold for repair of cartilage defects. The successful formation of new cartilaginous material in the biodendrimer-based hydrogel scaffold demonstrates the remarkable versatility and utility of the invention.
Dendritic Macromolecules
Dendritic polymers are globular monodispersed polymers composed of repeated branching units emanating from a central core. (US5714166; US4289872; US4435548;
US5041516; US5362843; US5154853; US05739256; US5602226; US5514764; all of which are explicitly incorporated by reference; Bosman, A. W.; Janssen, H. M.; Meijer, E.
W. Chem. Rev. 1999, 99, 1665-1688. Fischer, M.; Vogtle, F. Angew. Chem. Int. Ed. 1999,
38, 884-905. Zeng, F.; Zimmerman, S. C. Chem. Rev. 1997, 97, 1681-1712. Tomalia, D. A.; Naylor, A. M.; Goddard, W. A. Angew. Chem. Int. Ed. Engl. 1990, 29, 138.) These macromolecules are synthesized using either a divergent (from core to surface) (Buhleier,
W.; Wehner, F. V.; Vogtle, F. Synthesis 1987, 155-158. Tomalia, D. A.; Baker, H.;
Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Polymer Journal
1985, 17, 117-132. Tomalia, D. A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Macromolecules 1986, 19, 2466. Newkome, G. R.; Yao, Z.;
Baker, G. R.; Gupta, V. K. J. Org. Chem. 1985, 50, 2003.) or a convergent approach (from
surface to core). See Hawker, C. J.; Frechet, J. M. J. J. Am. Chem. Soc. 1990, 112, 7638-
7647. This research area has undergone tremendous growth in the last decade since the early work of Tomalia and Newkome. Compared to linear polymers, dendrimers are highly ordered, possess high surface area to volume ratios, and exhibit numerous end groups for functionalization. Consequently, dendrimers display several favorable physical properties for both industrial and biomedical applications including: small polydispersity indexes
(PDI), low viscosities, high solubility and miscibility, and excellent adhesive properties.
The majority of dendrimers investigated for biomedical/biotechnology applications (e.g.,
MRI, gene delivery, and cancer treatment) are derivatives of aromatic polyether or aliphatic amides, and thus are not ideal for in vivo uses. (Service, R. F. Science 1995, 267, 458-459.
Lindhorst, T. K.; Kieburg, C. Angew. Chem. Int. Ed. 1996, 35, 1953-1956. Ashton, P. R.;
Boyd, S. E.; Brown, C. L.; Yayaraman, N.; Stoddart, J. F. Angew. Chem. Int. Ed. 1997,
1997, 732-735. Wiener, E. C; Brechbeil, M. W.; Brothers, H.; Magin, R. L.; Gansow, O.
A.; Tomalia, D. A.; Lauterbur, P. C. Magn. Reson. Med. 1994, 31, 1-8. Wiener, E. C; Auteri, F. P.; Chen, J. W.; Brechbeil, M. W.; Gansow, O. A.; Schneider, D. S.; Beldford, R.
L.; Clarkson, R. B.; Lauterbur, P. C. J. Am. Chem. Soc. 1996, 775, 7774-7782. Toth, E.;
Pubanz, D.; Vauthey, S.; Helm, L.; Merbach, A. E. Chem. Eur. J. 1996, 2, 1607-1615.
Adam, G. A.; Neuerburg, J.; Spuntrup, E.; Muhl;er, A.; Scherer, K.; Gunther, R. W. J.
Magn. Reson. Imag. 1994, 4, 462-466. Bourne, M. W.; Margerun, L.; Hylton, N.; Campion, B.; Lai, J. J.; Dereugin, N.; Higgins, C. B. J. Magn. Reson. Imag. 1996, 6, 305-
310. Miller, A. D. Angew. Chem. Int. Ed. 1998, 37, 1768-1785. Kukowska-Latallo, J. F.;
Bielinska, A. U.; Johnson, J.; Spinder, R.; Tomalia, D. A.; Baker, J. R. Proc. Natl. Acad.
ScL 1996, 93, 4897-4902. Hawthorne, M. F. Angew. Chem. Int. Ed. 1993, 32, 950-984.
Qualmann, B.; Kessels M.M.; Musiol H.; Sierralta W.D.; Jungblut P.W.; L., M. Angew. Chem. Int. Ed. 1996, 35, 909-911).
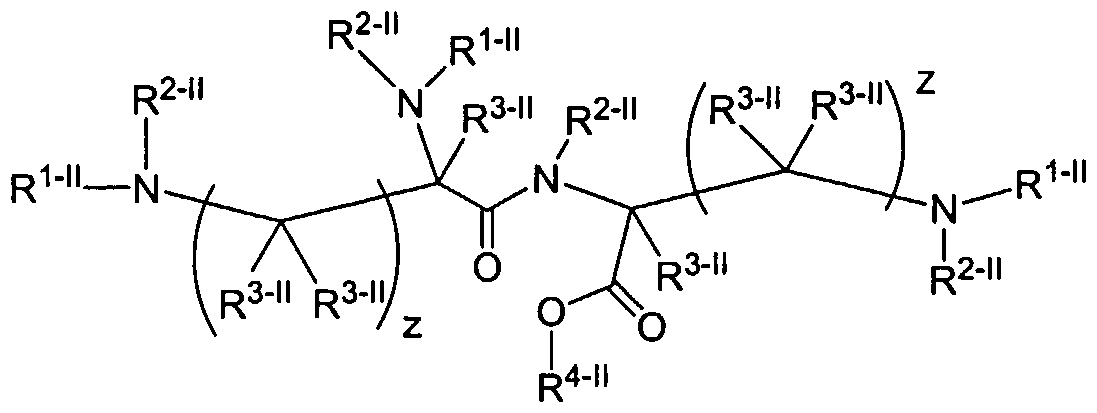
Biodendrimers are a novel class of dendritic macromolecules composed entirely of building blocks known to be biocompatible or degradable to natural metabolites in vivo. This application describes the synthesis, characterization, and use of novel dendrimers and dendritic macromolecules called "biodendrimers or biodendritic macromolecules" composed of such biocompatible or natural metabolite monomers such as but not limited to glycerol, lactic acid, glycolic acid, succinic acid, ribose, adipic acid, malic acid, glucose,
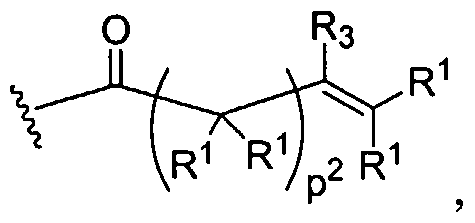
citric acid, glycine, lysine, cysteine, alanine, etc. A further embodiment of the invention is a dendritic structure that possess glycerol and one or more of lactic acid, glycolic acid, succinic acid, ribose, adipic acid, malic acid, glucose, citric acid, glycine, lysine, cysteine, alanine, etc. as a building block. In certain instances, the dendrimer is terminated with a photoreactive group or nucleophilic group. In certain instances, the terminus of the dendrimer contains a nucleoside. An additional embodiment of the invention is a dendritic structure that is composed of all lysine resides such that it is a generation one or higher or a lysine dendritic macromolecule terminated with cystene residues such that it is a generation one or higher. The present invention is generally in the area of the synthesis and fabrication of dendritic polymers and copolymers of polyesters, polyethers, polyether-esters, and polyamino acids or combinations thereof. For example, linear poly(glycolic acid), poly(lactic acid), and their copolymers are synthetic polyesters that have been approved by the FDA for certain uses, and have been used successfully as sutures, drug delivery carriers, and tissue engineering scaffold for organ failure or tissue loss (Gilding and Reed, Polymer, 20: 1459 (1979); Mooney et al., Cell Transpl., 2:203 (1994); and Lewis, D. H. in Biodegradable Polymers as Drug Delivery Systems, Chasin, M., and Langer, R., Eds., Marcel Dekker, New York, 1990). In tissue engineering applications, isolated cells or cell clusters are attached onto or embedded in a synthetic biodegradable polymer scaffold and this polymer-cell scaffold is next implanted into recipients (Langer and Vacanti, Science, 260:920 (1993). A large number of cell types have been used, including cartilage cells (Freed et al., Bio/Technology, 12:689 (1994)). Like the novel biodendrimers described in this invention, the advantages include their degradability in the physiological environment to yield naturally occurring metabolic products and the ability to control their rate of degradation by varying the ratio of lactic acid. In the dendritic structures, the degradation can be controlled by varying both the type of monomer used and the generation number.
A further embodiment of this invention is to attach biological recognition units for cell recognition to the end groups or within the dendrimer structure. For example, the tripeptide arginine-glycine-aspartic (RGD), can be added to the structure for cell binding. Barrera et al. described the synthesis of a poly(lactic acid) (pLAL) containing a low concentration of N-epsilon-carbobenzoxy-L-lysine units. The polymers were chemically
modified through reaction of the lysine units to introduce arginine-glycine-aspartic acid peptide sequences or other growth factors to improve polymer-cell interactions (Barrera et al., J. Am. Chem. Soc, 115:11010 (1993); U.S. Pat. No. 5,399,665 to Bartera et al.). The greatest limitation in the copolymers developed by Barrera et al. is that only a limited number of lysine units can be incorporated into the backbone. In many tissue engineering applications, the concentration of biologically active molecules attached to the linear polymer is too low to produce the desired interactions between the polymer and the body. Consequently, there is a need for the development of optimal materials for use as temporary scaffolds to support cell growth and tissue development in tissue engineering and wound repair applications. In addition, there is a need for methods for introducing functionalities such as polyamino acids, peptides, carbohydrates into polyesters, polyether-esters, polycarbonates, etc. in order to improve the biocompatibility, biochemical, mechanical, and other properties of the polymers. Furthermore, there is a need for the development of polyester, polyether ester, polyester-amines, etc. materials which include a sufficient concentration of derivatizable groups to permit the chemical modification of the polymer for different biomedical applications.
It is therefore an object of the invention to provide dendritic polymers and copolymers of polyesters and polyamino acids, polyethers, polyurethanes, polycarbonates, polycarbamates, polyamino alcohols or combinations of these polymer classess which can be chemically modified for different biomedical applications such as tissue engineering applications, wound management, contrast agents vehicles, drug delivery vechiles, etc. It is a further object of the invention to provide dendritic polymers and copolymers of polyesters and polyamino acids with improved properties such as biodegradability, biocompatibility, mechanical strength. It is still another object of the invention to provide dendritic polymers that can be derivatized to include functionalities such as peptide sequences or growth factors to improve the interaction of the polymer with cells, tissues, or bone.
The advantages of a dendritic polymer include multiple end groups for functionalization, crosslinked gels with high crosslinking densities at low polymer concentration, globular structure, low viscosities, and well-defined composition. Conventional linear polymers for medical applications cannot be easily controlled or modified through changes in the polymer's structure, because these polymers (e.g., PLA) do
not possess functional groups, other than end groups, that permit chemical modification to change their properties, and these polymers do not adopt a well-defined structure in solution, thereby limiting the applications of these polymers. Consequently the novel polymers described herein are substantially different.
Gels
Another aspect of the present invention relates to using dendritic polymeric gels, gel-cell, gel-drug compositions for orthopedic surgeries, drug delivery, and tissue engineering. Gels are 3D polymeric materials which exhibit the ability to swell in water and to retain a fraction of water within the structure without dissolving. The physical properties exhibited by gels such as water content, sensitivity to environmental conditions {e.g., pH, temperature, solvent, stress), softness, adhesivity, and rubbery consistency are favorable for biomedical and biotechnological applications. Indeed, gels may be used as coatings {e.g. biosensors, catheters, and sutures), as "homogeneous" materials {e.g. contact lenses, burn dressings, and dentures), and as devices (e.g. artificial organs and drug delivery systems) (Peppas, N. A. Hydrogel in Medicine and Pharmacy, VoI I and II 1987. Wichterle, O.; Lim, D. Nature 1960, 185, 117-118. Ottenbrite, R. M.; Huang, S. J.; Park, K. Hydrogels and Biodegradable polymers for Bioapplications 1994; Vol. 627, pp 268).
For example, gel matrices for the entrapment of cells, including stem cells, as artificial organs/tissues have been explored for more than fifteen years in some applications, and encapsulation is a promising approach for a number of disease states including Parkinson's disease (L-dopamine cells), liver disease (hepatocyte cells), and diabetes (islets of Langerhans). In the past, for example, islets of Langerhans (the insulin producing cells of the pancreas) have been encapsulated in an ionically crosslinked alginate (a natural hydrogel) microcapsule with a poly-L-lysine coating, and successfully reduced blood sugar levels in diabetic mice following transplantation.
Another aspect of the present invention relates to a method and means for designing, constructing, and utilizing artificial dendritic matrices as temporary scaffolding for cellular growth and implantation. A further embodiment of the invention to provide biodegradable, non-toxic matrices which can be utilized for cell growth, both in vitro, in vivo, and in situ. The cell scaffold/matrix/gel can be formed in vitro or in situ by crosslinking. It is another
object of the present invention to provide a method for configuring and constructing biodegradable artificial matrices such that they not only provide a support for cell growth, but allow and enhance vascularization and differentiation of the growing cell mass following implantation. It is yet another object of the invention to provide matrices in different configurations so that cell behavior and interaction with other cells, cell substrates, and molecular signals can be studied in vitro.
Biologically Active Agents Within the Dendritic Gel/Network
In certain instances, biologically active agents may be incorporated in the the dendritic gel. Active agents amenable for use in the compositions of the present invention include growth factors, such as transforming growth factors (TGFs), fibroblast growth factors (FGFs), platelet derived growth factors (PDGFs), epidermal growth factors (EGFs), connective tissue ctivated peptides (CTAPs), osteogenic factors, and biologically active analogs, fragments, and derivatives of such growth factors. Members of the transforming growth factor (TGF) supergene family, which are multifunctional regulatory proteins, are particularly preferred. Members of the TGF supergene family include the beta transforming growth factors (for example, TGF-βl, TGF-β2, TGF-β3); bone morphogenetic proteins (for example, BMP-I, BMP-2, BMP-3, BMP-4, BMP-5, BMP-6, BMP-7, BMP-8, BMP-9); heparin-binding growth factors (for example, fibroblast growth factor (FGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF)); Inhibins (for example, Inhibin A, Inhibin B); growth differentiating factors (for example, GDF- 1); and Activins (for example, Activin A, Activin B, Activin AB).
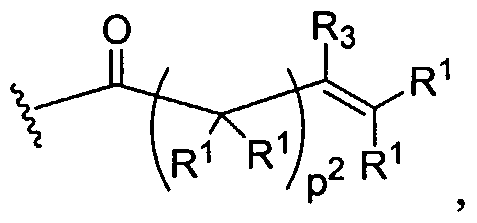
Crosslinked Gels or Networks To prepare the dendritic crosslinked gel/network of the present invention, dendrimers or dendritic polymers are crosslinked using either light or a chemical crosslinking reaction that is not activated by light. A large number of crosslinking reactions are amenable to the present invention. For example, the crosslinking reaction may be an acyrylate polymerization initiated by light, reaction of a dihydrazide with a diketone to make a stablized imine, a siloxane crosslinking reaction, or a nucleophilic attack onto an electrophilic site such as reaction of a thiol or amine with an activated ester, aldol
condensation, and the like. A further embodiment of this invention is the crosslinking between dendritic polymers and dendritic polymers and linear polymers or any combination thereof to form a crosslinked gel or network. The gels can be highly hydrated and hydrophilic; such gels are often called hydrogels. For the chemical crosslinking reaction that is not light-activated, the polymers are functionalized to contain groups that will react with each other to form the gel. For example, the dendritic polymers have been chemically modified to have more than two functional groups such nucleophilic groups, such as primary amino (-NH2) groups or thiol (-SH) groups, which can react with electrophilic groups such as an acrylate, succinimidyl ester, maleimide, or aldehyde. Each functional group on a multifunctionally dendritic polymer is capable of covalently binding with another polymer, thereby effecting crosslinking between the polymers and formation of the network.
Examples of covalently crosslinked networks can be formed by reacting an activated ester (such as a succinimidyl ester) with an amine or thiol (such as a terminal primary or secondary amine, lys, cys, etc.) Thiol or cysteine terminated dendritic structure that forms a disulfide crosslinked network with another thiol or cysteine terminated dendritic(s) or linear polymer(s) will also form a gel. Alternatively, a gel is formed during the reaction of an aldehyde-functionalized small molecule or polymer and an amine- or cysteine-functionalized polymer. An additional method is to have a maleimide- or vinylsulfone-functionalized dendritic polymer react with a thiol-functionalized dendritic, linear, comb, or other polymer to form the gel. A functionalized succinimidyl glutarate dendritic polymer with an acid-terminated dendritic, linear, comb, or other polymer to from the gel. An acrylate-functionalized polymer reacts with an amine- or thiol-functionalized polymer to form the crosslinked gel. A further embodiment of this invention is the use of a chemical peptide ligation reaction to create a crosslinked gel involving a dendritic polymer. In this reaction an aldehyde or aldehyde-acid reacts with a cysteine-functionalized polymer to form a gel or crosslinked network.
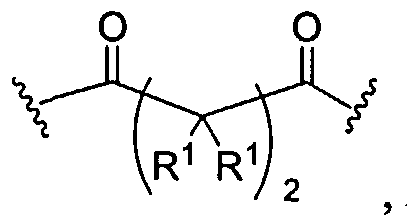
Biodendrimers based on a core unit and branches composed of glycerol and lactic acid, glycerol and glycolic acid, glycerol and succinic acid, glycerol and adipic acid, and glycerol, succinic acid, and PEG represent examples of this class of polymers, according to the present invention. Thus, one can build a wide range of structures as shown below.
After the core is synthesized, polymers such as PEG and PLA can be attached to the core unit or to a branch to make large starburst or dendritic polymers.
The gels of the invention can be formed by applying a dendrimeric compound to a cartilege defect of a patient, and then exposing the dendrimeric compound to a polymerization agent. For example, a dendrimeric compound having acrylate groups attached to the periphery of the dendrimer is applied to a cartilege defect of a patient, and then the dendrimeric compound is exposed to ultraviolet radiation. In certain instances, a dendrimeric compound having a nucleophilic group attached to the periphery of the dendrimer is applied to a cartilege defect of a patient, and then the dendrimeric compound is exposed to a compound having electrophilic groups.
Alternatively, a polymerization agent is applied to a cartilege defect of a patient, and then the polyermization agent is exposed to a dendrimeric compound. For example, PEG(NHS)2 is applied to a cartilege defect of a patient, and then PEG(NHS)2 is exposed to a dendrimeric compound having a nucleophilic group attached to the periphery of the dendrimer.
Notably, the polymerization agent may be a copolymer containing either nucleophilic or electrophilic endgroups. A large number of copolymers are known the art and are amenable to the present invention. In certain instances, the copolymer comprises hydrophobic and hydrophilic domains. In certain instances, the polymerization agent is a copolymer of polyethylene glycol and polypropylene glycol, wherein the copolymer has either nucleophilic or electrophilic endgroups attached to the ends of the copolymer.
Cartilaginous Tissue
Cartilage is a tough, flexible, elastic biomaterial that serves as flexible connective tissue commonly found covering the surface on many joints in animals. Cartilage serves to reduce friction between bones and absorb shocks due to sudden increases in the amount of weight applied to the skeletal system. Cartilage is made of chondrocytes and chondroblasts dispersed in a lipoprotein that is reinforced with collagen fibers. Cartilage is located in many parts of the human body. For example, cartilage is found in the tip of the nose, in the ribs, in the external portion of the ear, walls of the trachea, and covering the surface of bones where joints occur. The three main types of cartilage are articular (hyaline),
fibrocartilage, and elastic cartilage. Articular cartilage, also known as hyaline cartilage, is present in the human body on the ends of bones that form joints and on the ends of ribs. Representative examples of joints containing cartilage tissue include knee, hip, ankle, elbow, wrist, shoulder, fingers, toes, spinal column, and the like. Fibrocartilage contains a substantial amount of collagen. Fibrocartilage is located between bones in the spinal column, hip, and pelvis. The meniscus is a type of fibrocartilage that can be found covering bone tissue in the knee. Elastic cartilage can be found in the outer ear and epiglottis.
A number of defects are known to occur in cartilage tissue. One of the more common defects that occurs to cartilage tissue is tearing. Tearing of cartilage tissue is a common knee injury that often requires surgery and extension physical therapy in order to recover. Another common cartilage defect is when cartilage tissue simply deteriorates due to prolonged wear and tear. This type of cartilage defect is more common in those patients that have performed hard physical labor over a period of many years. Activities such as heavy lifting or repetitive motions can accentuate the rate at which cartilage tissue deteriorates. In certain instances, deterioration of the cartilage tissue causes the layer of cartilage tissue protecting the bone at a joint to become too thin for sufficient protection. Other types of cartilege defects include cracking, fibrillation, strains, and rough cartilage surfaces. The size of the defect can vary considerably. In certain instances, the defect can apply to nearly the entire portion of the cartilage tissue that covers the bone tissue of a certain joint. In other instances, the defective cartilage tissue is located on only a small, localized portion of the cartilage tissue covering a joint. In certain instances, the cartilage tissue defect is less than about 15 cm . In certain instances, the cartilage tissue defect is less than about 10 cm2. In certain instances, the cartilage tissue defect is less than about 5 cm2. In certain instances, the cartilage tissue defect is less than about 2 cm2. In certain instances, the cartilage tissue defect is less than about 1 cm2. In certain instances, the cartilage tissue defect is less than about 0.5 cm2. In certain instances, the cartilage tissue defect is a tear in the cartilage tissue. In certain instances, the cartilage tissue defect is less than about 3 cm long. In certain instances, the cartilage tissue defect is less than about 2 cm long. In certain instances, the cartilage tissue defect is less than about 1 cm long. In certain instances, the cartilage tissue defect is less than about 0.5 cm long.
Certain Embodiments of the Invention
The present invention is described below by reference to specific embodiments. This description is not meant to limit the scope of the invention, but to convey the essence of the invention. Additional embodients may be readily envisioned by one of ordinary skill in the art, and such embodiments fall within the scope of the invention.
One aspect of the present invention relates to a method for preparing and administrating in situ a biocompatible gel ex vivo, in vitro, or in vivo, comprising: (a) forming a reactive composition by admixing a biocompatible crosslinking polymer having two different nucleophilic groups such as sulfhydryl and amine groups where there is at least one amine or sulfhydryl group on the polymer with a biocompatible crosslinking polymer B having amine and sulfhydryl-reactive groups, and further wherein the amine and sulfhydryl-reactive groups are capable of covalent reaction with the amine and sulfhydryl groups upon admixture of polymers A and B under effective crosslinking conditions to form a gel in less than one day; and (b) allowing the components of the reactive composition to crosslink and thereby form a gel.
Another aspect of the present invention relates to dendritic or branched polymers or copolymers composed of monomers synthesized by combining branching compounds with other linear or branched building blocks. Both components are known to be biocompatible or are natural metabolites in vivo including but not limited to glycerol, citric acid, lactic acid, glycolic acid, adipic acid, caproic acid, ribose, glucose, succinic acid, malic acid, amino acids, peptides, synthetic peptide analogs, poly(ethylene glycol), poly(hydroxyacids) [e.g., PGA. PLA], including where one of the monomers is a branched structure such as glycerol combined with one of the other components. In certain instances, the present invention relates to the aforementioned polymers derivatized with peripheral compounds possessing an olefin including but not limited to acrylate, methacrylate.
In certain instances, the present invention relates to the the aforementioned polymers derivatized with peripheral compounds including but not limited to cysteine, lysine, other amino acids, or any other compounds that would provide terminal nucleophiles
(including but not limited to amines, thiols, hydroxyl groups) or electrophiles (including but not limited to NHS esters, maleimides, aldehydes, ketones).
In certain instances, the present invention relates to the the aforementioned polymers for subsequent polymerization/crosslinking/reaction with another linear or branched structure with either olefϊnic, electrophilic or nucleophilic groups, respectively to form a gel.
In certain instances, the present invention relates to the the aforementioned polymers for subsequent polymerization/crosslinking/reaction with another linear or branched structure via a photopolymerization process (single or multi-photon process) to form a gel.
Another aspect of the present invention relates to a branching structure with at least three functional groups composed of but not limited to glycerol, citric acid, malic acid, amino acids, peptides, synthetic peptide analogs, or other dendritic strucutures synthesized to produce terminal olefins (including but not limited to acrylate or methacrylate groups), nucleophiles (including but not limited to amines, thiols, hydroxyl groups) or electrophiles (including but not limited to NHS esters, maleimides, aldehydes, ketones) for subsequent polymerization/crosslinking with another linear or branched structure with either olefϊnic, electrophilic or nucleophilic groups, respectively.
Another aspect of the present invention relates to a branching structure with at least three functional groups composed of but not limited to glycerol, citric acid, malic acid, amino acids, peptides, synthetic peptide analogs, or other dendritic structures derivatized with peripheral compounds including but not limited to cysteine, lysine, other amino acids, or any other compounds that would provide terminal olefins (including but not limited to acrylate or methacrylate groups), nucleophiles (including but not limited to amines, thiols, hydroxyl groups) or electrophiles (including but not limited to NHS esters, maleimides, aldehydes, ketones) for subsequent polymerization/crosslinking with another linear or branched structure with either olefinic, electrophilic or nucleophilic groups, respectively.
Another aspect of the present invention relates to a branching structure composed of three lysine amino acids with four cysteine amino acids on the periphery with the structure CysLys(Cys)Lys(CysLys(Cys))OMe«4HCl as described in the examples.
Another aspect of the present invention relates to a branching structure composed of three lysine amino acids with amines on the periphery with the structure (Lys)Lys(Lys)OMe»4HCl as described in the examples.
In certain instances, the present invention relates to the aforementioned polymers for subsequent polymerization/crosslinking/reaction with another linear or branched structure with olefϊnic, electrophilic or nucleophilic groups to form a gel.
In certain instances, the present invention relates to the aforementioned polymers for subsequent polymerization/crosslinking/reaction with another linear or branched structure through thiazolidine linkages to form a gel. In certain instances, the present invention relates to the aforementioned polymers undergoing polymerization/crosslinking with a poly(ethylene glycol) molecular weight of about 200 to about 200,000 with at least two electrophilic groups.
In certain instances, the present invention relates to the aforementioned polymers undergoing polymerization/crosslinking with a poly(ethylene glycol) molecular weight of about 200 to about 200,000 with at least two nucleophilic groups
In certain instances, the present invention relates to the aforementioned polymers undergoing polymerization/crosslinking with a poly(ethylene glycol) molecular weight of about 200 to about 200,000 with functional groups including but not limited to olefins, aldehydes, maleimides, or NHS esters. In certain instances, the present invention relates to the aforementioned polymers undergoing polymerization/crosslinking with a poly(ethylene glycol) molecular weight of about 200 to about 200,000 with aldehyde functional groups to form hydrogels through the formation of thiazolidine linkages.
In certain instances, the present invention relates to the the aforementioned formulations in which each of the components are dissolved or suspended in an aqueous solution wherein the said aqueous solution is selected from water, buffered aqueous media, saline, buffered saline, solutions of amino acids, solutions of sugars, solutions of vitamins, solutions of carbohydrates or combinations of any two or more thereof.
In certain instances, the present invention relates to the application of the aforementioned formulation through a delivery device which physically separates the
components until the components are physically mixed by the end user, including but not limited to a dual barrel syringe with a mixing device.
Another aspect of the present invention relates to packaging of the aforementioned branching compounds in an aqueous solution at a preselected pH and molarity selected from the aqueous solutions described above and the packaging of the second compound in an aqueous solution at another preselected pH and molarity selected from the aqueous solutions described above. When combined, the pH and molarities of the two solutions produce a final desired solution with a different pH.
Another aspect of the present invention relates to packaging of the aforementioned branching compounds in an aqueous solution at a preselected pH and molarity selected from the aqueous solutions described above and the packaging of the second compound in an aqueous solution at another preselected pH and molarity selected from the aqueous solutions described above. The contents are packaged free of oxygen and shielded from light. When combined, the pH and molarities of the two solutions produce a final desired solution with a different pH.
Another aspect of the present invention relates to packaging of the aforementioned branching compounds as a powder and adding an aqueous solution at a preselected pH and molarity selected from the aqueous solutions described above before use. The second component may either be packaged by dissolving the second compound in an aqueous solution at another preselected pH and molarity selected from the aqueous solutions described above or packaged similar to the first compound in which the compound stored as a powder and an aqueous solution at a preselected pH and molarity selected from the aqueous solutions described above is added before use. The contents are packaged free of oxygen and shielded from light. When combined, the pH and molarities of the two solutions produce a final desired solution with a different pH.
Another aspect of the present invention relates to the storage of the aforementioned cystein terminated polymers in an acidic, oxygen free solution to minimize the formation of disulfide bonds.
Another aspect of the present invention relates to the storage of the aforementioned aldehyde terminated polymers in an acidic, oxygen free solution to maximize the percent reactivity of the polymer and minimize aldol condensation and reverse Michael additions.
Another aspect of the present invention relates to the addition of various additives that might be incorporated into the polymer formulations including, but not limited to, antioxidants, colorants, viscosity modifiers, plasticizers, small molecule carbohydrates, large molecule carbohydrates, amino acids, peptides, or other water soluble polymers (linear or branched). Such additives may be added to increase the shelf life, increase the polymerization rate, modifϊy the pH or molarity of the solution, change the refractive index, modify the mechanical properties, change crosslinking density, decrease swelling, or aid in visualization.
Another aspect of the present invention relates to the addition of various additives or anti-microbial agents such has polyhexamethylene biguanide (PHMB) that might be incorporated into the polymer formulations.
Another aspect of the present invention relates to the resulting hydrogels formed by mixing the aforementioned compounds as described and prepared above.
In certain instances, the present invention relates to hydrogels formed by photopolymerization of the aforementioned compounds.
Another aspect of the present invention relates to a method of using crosslinkable/polymerizable/reactionary dendritic polymers, branching structures, and their hydrogels for delivery of therapeutics.
Another aspect of the present invention relates to a method of using a crosslinkable/polymerizable/reactionary dendritic polymer or monomer for seeding with cells and subsequent in situ polymerization in vivo.
Another aspect of the present invention relates to a method of using a crosslinkable/polymerizable/reactionary branched or dendritic polymer for drug delivery.
Another aspect of the present invention relates to a crosslinkable/polymerizable/reactionary dendritic polymer or monomer wherein the crosslinking is of covalent, ionic, electrostatic, and/or hydrophobic nature.
Another aspect of the present invention relates to a crosslinkable dendritic polymer or monomer wherein the crosslinking reaction involves a nucleophile and electrophile.
Another aspect of the present invention relates to a crosslinkable dendritic polymer or monomer wherein the crosslinking reaction is a peptide ligation reaction.
Another aspect of the present invention relates to a crosslinkable dendritic polymer or monomer wherein the crosslinking reaction is a Diels-Alder reaction.
Another aspect of the present invention relates to a crosslinkable dendritic polymer or monomer wherein the crosslinking reaction is a Michael Addition reaction. Another aspect of the present invention relates to a crosslinkable dendritic polymer or monomer wherein the crosslinking reaction is a photochemical reaction using a UV or visible photoinitator chromophore.
Another aspect of the present invention relates to a crosslinkable branched or dendritic polymer in combination with a crosslinkable linear, comb, multi-block, star, or dendritic polymer(s) for a medical or tissue engineering application.
Another aspect of the present invention relates to a crosslinkable branched or dendritic polymer in combinaton with a crosslinkable monomer(s) for a medical or tissue engineering application.
Another aspect of the present invention relates to a method of using a crosslinkable branched or dendritic polymer combined with a crosslinkable small molecule(s) (molecule weight less than about 1000 daltons) for a medical or tissue engineering application.
Another aspect of the present invention relates to a crosslinkable branched or dendritic polymer or monomer wherein the said crosslinking dendritic polymer is combined with one or more linear, comb, multi-block, star polymers or crosslinkable comb, multi- block, star polymers.
Another aspect of the present invention relates to a crosslinkable dendritic polymer or monomer wherein the final polymeric form is a gel, film, fiber, or woven sheet.
Another aspect of the present invention relates to the aforementioned polymers, branching structures, and their resulting hydrogels wherein the final polymeric form is a gel, film, fiber, or woven sheet.
Another aspect of the present invention relates to the aforementioned polymers, branching structures, and their resulting hydrogels wherein the polymer or crosslinkable monomer is D or L configuration or a mixture.
Another aspect of the present invention relates to the aforementioned polymers, branching structures, and their hydrogels wherein the dendritic structure is asymmetric at the surface such as a surface block structure where a carboxylate acid(s) and alkyl chains,
or acrylate(s) and PEG(s) are present, for example, or within the core and inner layers of the dendrimer such as amide and ester linkages in the structure.
Another aspect of the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer wherein the polymer is a star biodendritic polymer or copolymer as shown in at least one of the formulas below: where Y and X are the same or different at each occurrence and are O, S, Se, N(H), or P(H) and where Ri, R2, R3, R4, Rs, R6, R7, R8, A or Z are the same or different and include -H, -CH3, -OH, carboxylic acid, sulfate, phosphate, aldehyde, methoxy, amine, amide, thiol, disulfide, straight or branched chain alkane, straight or branched chain alkene, straight or branched chain ester, straight or branched chain ether, straight or branched chain silane, straight or branched chain urethane, straight or branched chain, carbonate, straight or branched chain sulfate, straight or branched chain phosphate, straight or branched chain thiol urethane, straight or branched chain amine, straight or branched chain thiol urea, straight or branched chain thiol ether, straight or branched chain thiol ester, or any combination thereof.
Another aspect of the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer where the straight or branched chain is of about 1-50 carbon atoms wherein the chain is fully saturated, fully unsaturated or any combination therein
In certain instances, the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer where the straight or branched chain is of about 1-50 carbon atoms wherein the chain is fully saturated, fully unsaturated or any combination therein.
In certain instances, the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer wherein straight or branched chains are the same number of carbons or different wherein Ri, R2, R3, R4, Rs, R6, R7, R8, A or Z are any combination of the linkers including ester, silane, urea, amide, amine, carbamate, urethane, thiol-urethane, carbonate, thio-ether, thio-ester, sulfate, phosphate and ether.
In certain instances, the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer which includes at least one chain selected from the group consisting of hydrocarbons, flourocarbons, halocarbons, alkenes, and alkynes.
In certain instances, the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer which includes at least one chain selected from the group consisting of linear and dendritic polymers.
In certain instances, the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer wherein said linear and dendritic polymers include at least one selected from the group consisting of polyethers, polyesters, polyamines, polyacrylic acids, polycarbonates, polyamino acids, polynucleic acids and polysaccharides of molecular weight ranging from about 200-1,000,000, and wherein said chain contains 0, 1 or more than 1 photopolymerizable group.
Another aspect of the present invention relates to a crosslinkable or noncrosslinkable polymer, wherein the polyether is PEG, and wherein the polyester is PLA, PGA or PLGA.
Another aspect of the present invention relates to a linear polymer wherein the chain is a polymer or copolymer of a polyester, polyamide, polyether, or polycarbonate of or the aforementioned polymer in combination with a polyester, polyamide, polyether, or polycarbonate of:
R5 R6
-H-j R3-VJ A-j R2-xJ-lr- t
Structure I
Structure III
In certain instances, the present invention relates to the aforementioned polymer comprised of repeating units of general Structure I, where A is O, S, Se, or N-R7. In certain instances, the present invention relates to the aforementioned polymer, where W, X, and Z are the same or different at each occurrence and are O, S, Se, N(H), or P(H).
In certain instances, the present invention relates to the aforementioned polymer, where Ri is hydrogen, a straight or branched alkyl chain of about 1 -20 carbons, cycloalkyl, aryl, olefin, silyl, alkylsilyl, arylsilyl, alkylaryl, or arylalkyl group.
In certain instances, the present invention relates to the aforementioned polymer, where Ri is hydrogen, a straight or branched alkyl chain of about 1-20 carbons, cycloalkyl, aryl, olefin, silyl, alkylsilyl, arylsilyl, alkylaryl, or arylalkyl group substituted internally or terminally by one or more hydroxyl, hydroxyether, carboxyl, carboxyester, carboxyamide, amino, mono- or di-substituted amino, thiol, thioester, sulfate, phosphate, phosphonate, or halogen substituents.
In certain instances, the present invention relates to the aforementioned polymer, where Ri is a polymer (such as poly(ethylene glycol), poly(ethylene oxide), or a poly(hydroxyacid)), a carbohydrate, a protein, a polypeptide, an amino acid, a nucleic acid, a nucleotide, a polynucleotide, any DNA or RNA segment, a lipid, a polysaccharide, an antibody, a pharmaceutical agent, or any epitope for a biological receptor.
In certain instances, the present invention relates to the aforementioned polymer, where Ri is a photocrosslinkable, chemically, or ionically crosslinkable group.
In certain instances, the present invention relates to the aforementioned polymer, in which D is a straight or branched alkyl chain of about 1-5 carbons, m is 0 or 1, and R2, R3,
R4, R5, R6, and R7 are the same or different at each occurrence and are hydrogen, a straight or branched alkyl chain of about 1-20 carbons, cycloalkyl, aryl, alkoxy, aryloxy, olefin,
alkylamine, dialkylamine, arylamine, diarylamine, alkylamide, dialkylamide, arylamide, diarylamide, alkylaiyl, or arylalkyl group.
In certain instances, the present invention relates to the aforementioned polymer comprised of repeating units of General Structure II, where L, N, and J are the same or different at each occurrence and are O, S, Se, N(H), or P(H).
In certain instances, the present invention relates to the aforementioned polymer where Ri is hydrogen, a straight or branched alkyl chain of about 1 -20 carbons, cycloalkyl, aryl, olefin, silyl, alkylsilyl, arylsilyl, alkylaryl, or arylalkyl group.
In certain instances, the present invention relates to the aforementioned polymer where Ri is hydrogen, a straight or branched alkyl chain of about 1-20 carbons, cycloalkyl, aryl, olefin, silyl, alkylsilyl, arylsilyl, alkylaryl, or arylalkyl group substituted internally or terminally by one or more hydroxyl, hydroxyether, carboxyl, carboxyester, carboxyamide, amino, mono- or di-substituted amino, thiol, thioester, sulfate, phosphate, phosphonate, or halogen substituents. In certain instances, the present invention relates to the aforementioned polymer where Ri is a polymer selected from the group consisting of poly(ethylene glycols), poly(ethylene oxides), and poly(hydroxyacids, or is a carbohydrate, a protein, a polypeptide, an amino acid, a nucleic acid, a nucleotide, a polynucleotide, a DNA or RNA segment, a lipid, a polysaccharide, an antibody, a pharmaceutical agent, or an epitope for a biological receptor.
In certain instances, the present invention relates to the aforementioned polymer where Ri is a photocrosslinkable, chemically, or ionically crosslinkable group.
In certain instances, the present invention relates to the aforementioned polymer, where D is a straight or branched alkyl chain of about 1-5 carbons, q and r are the same or different at each occurrence and are 0 or 1 , and R7, R8, R9, R10, Rn, R12, R13, and Ri4 are the same or different at each occurrence and are hydrogen, a straight or branched alkyl chain of about 1-20 carbons, cycloalkyl, aryl, alkoxy, aryloxy, olefin, alkylamine, dialkylamine, arylamine, diarylamine, alkylamide, dialkylamide, arylamide, diarylamide, alkylaryl, or arylalkyl group. In certain instances, the present invention relates to the aforementioned block or random copolymer comprised of repeating units of general Structure III, where M, T, and Q
are the same or different at each occurrence and are O, S, Se, N(H), or P(H), e is 0 or 1-9, and Ri 5 is a straight or branched alkyl chain of about 1-5 carbons, unsubstituted or substituted with one or more hydroxyl, hydroxyether, carboxyl, carboxyester, carboxyamide, amino, mono- or di-substituted amino, thiol, thioester, sulfate, phosphate, phosphonate, or halogen substituents
In certain instances, the present invention relates to the aforementioned block or random copolymer comprised of repeating units of general Structure III, where M, T, and Q are the same or different at each occurrence and are O, S, Se, N(H), or P(H), and R15 is a straight or branched alkyl chain of about 1-5 carbons, unsubstituted or substituted with one or more hydroxyl, hydroxyether, carboxyl, carboxyester, carboxyamide, amino, mono- or di-substituted amino, thiol, thioester, sulfate, phosphate, phosphonate, or halogen substituents.
In certain instances, the present invention relates to the aforementioned block or random copolymer comprised of repeating units of general Structure III, where M, T, and Q are the same or different at each occurrence and are O, S, Se, N(H), or P(H), and Rl 5 is a straight or branched alkyl chain of about 1-5 carbons, unsubstituted or substituted with one or more hydroxyl, hydroxyether, carboxyl, carboxyester, carboxyamide, amino, mono- or di-substituted amino, thiol, thioester, sulfate, phosphate, phosphonate, or halogen substituents. Another aspect of the present invention relates to a higher order block or random copolymer comprised of three or more different repeating units, and having one or more repeating units described above, such as a polyglyerol glycine carbonate-polyglycerol succinic acid copolymer.
Another aspect of the present invention relates to a block or random copolymer as described above, which includes at least one terminal crosslinkable group selected from the group consisting of amines, thiols, amides, phosphates, sulphates, hydroxides, alkenes, and alkynes.
In certain instances, the present invention relates to the aforementioned block or random copolymer where X, Y, M is O, S, N-H, N-R, and wherein R is -H, CH2, CR2, Se or an isoelectronic species of oxygen.
In certain instances, the present invention relates to the aforementioned block or random copolymer wherein an amino acid(s) is attached to R], R2, R3, R4, R5, A, and/or Z.
In certain instances, the present invention relates to the aforementioned block or random copolymer wherein a polypeptide(s) is attached to Ri, R2, R3, R4, Rs, A, and/or Z. In certain instances, the present invention relates to the aforementioned block or random copolymer wherein an antibody(ies) is attached to Ri, R2, R3, R4, R5, A, and/or Z.
In certain instances, the present invention relates to the aforementioned block or random copolymer wherein a nucleotide(s) is attached to Ri, R2, R3, R4, R5 A, and/or Z.
In certain instances, the present invention relates to the aforementioned block or random copolymer wherein a nucleoside(s) is attached to Ri, R2, R3, R4, R5, A, and/or Z.
In certain instances, the present invention relates to the aforementioned block or random copolymer wherein an oligonucleotide(s) is attached to Ri, R2, R3, R4, R5, A, and/or Z.
In certain instances, the present invention relates to the aforementioned block or random copolymer wherein a ligand(s) is attached to Ri, R2, R3, R4, Rs, A, and/or Z that binds to a biological receptor.
In certain instances, the present invention relates to the aforementioned block or random copolymer wherein a pharmaceutical agent(s) is attached to Ri, R2, R3, R4, R5, A, and/or Z. In certain instances, the present invention relates to the aforementioned crosslinkable or noncrosslinkable polymer or copolymer wherein the polymer is a dendritic macromolecule including at least one polymer selected from the group consisting of dendrimers, hybrid linear-dendrimers, dendrons, or hyperbranched polymers according to one of the general formulas or such similar structures below: where R3, R4, which may be the same or different, are a repeat pattern of B, and n is about 0 to 50.
41
In certain instances, the present invention relates to the aforementioned polymer, wherein X, Y, M is O, S, N-H, N-R, wherein R is -H, CH2, CR2 or a chain as defined above, Se or any isoelectronic species of oxygen
In certain instances, the present invention relates to the aforementioned polymer, wherein X, Y, M is O, S, N-H, N-R, wherein R is -H, CH2, CR2 or a chain as defined above, Se or any isoelectronic species of oxygen.
In certain instances, the present invention relates to the aforementioned polymer where R3 and R4 are carboxylic acid with a protecting group such as but not limited to a phthalimidomethyl ester, a t-butyldimethylsilyl ester, or a t-butyldiphenylsilyl ester.
In certain instances, the present invention relates to the aforementioned polymer where R3, R4, A, and Z are the same or different, R3 and R4 are repeated a certain number of times, and terminate in -H, -OH, -CH3, carboxylic acid, sulfate, phosphate, aldehyde, activated ester, methoxy, amine, amide, thiol, disulfide, straight or branched chain alkane, straight or branched chain alkene, straight or branched chain ester, straight or branched chain ether, straight or branched chain silane, straight or branched chain urethane, straight or branched chain, carbonate, straight or branched chain sulfate, straight or branched chain
phosphate, straight or branched chain thiol urethane, straight or branched chain amine, straight or branched chain thiol urea, straight or branched chain thiol ether, straight or branched chain thiol ester, or any combination thereof, and wherein c is a natural or un¬ natural amino acid. In certain instances, the present invention relates to the aforementioned polymer having a straight or branched chain of 1-50 carbon atoms and wherein the chain is fully saturated, fully unsaturated or any combination therein.
In certain instances, the present invention relates to the aforementioned polymer wherein straight or branched chains are the same number of carbons or different and wherein R3, R4, A, Z are any combination of linkers selected from the group consisting of esters, silanes, ureas, amides, amines, urethanes, thiol-urethanes, carbonates, carbamates, thio-ethers, thio-esters, sulfates, phosphates and ethers.
In certain instances, the present invention relates to the aforementioned polymer wherein chains include at least one selected from hydrocarbons, flourocarbons, halocarbons, alkenes, and alkynes.
In certain instances, the present invention relates to the aforementioned polymer wherein said chains include polyethers, polyesters, polyamines, polyacrylic acids, polyamino acids, polynucleic acids and polysaccharides of molecular weight ranging from
200-1,000,000, and wherein said chain contains 1 or more crosslinkable or photopolymerizable group.
In certain instances, the present invention relates to the aforementioned polymer wherein the chains include at least one of PEG, PLA, PGA, PGLA, and PMMA.
In certain instances, the present invention relates to the aforementioned block or random copolymer, which includes at least one terminal crosslinkable or photopolymerizable group selected from the group consisting of amines, thiols, amides, phosphates, sulphates, hydroxides, alkenes, activated esters, malemides, aldehydes, and alkynes.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with amino acid(s), such as cysteine, attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with polypeptide(s) attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with an antibody(ies) or single chain antibody(ies) attached to Z, A, R3, and/or R4. In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with a nucleotide(s) attached to Z, A, R3, and/or R4..
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with a nucleoside(s) attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with oligonucleotide(s) attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with ligand(s) attached to Z, A, R3, and/or R4 that binds to a biological receptor.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with a pharmaceutical agent(s) attached to Z, A, R3, and/or R4. In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with a pharmaceutical agent attached to Z, A, R3, and/or R4 and is at least one selected from the group consisting of antibacterial, anticancer, anti-inflammatory, and antiviral.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times to produce a polymer in which a pharmaceutical agent(s) is encapsulated or chemically bound to the polymer.
In certain instances, the present invention relates to the aforementioned polymer wherein camptothecin or a deriviative of campothethcin is encapsulated
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with a carbohydrate(s) attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with a PET or MRI contrast agent(s) attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein the contrast agent is Gd(DPTA).
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with an iodated compound for X-ray imagaging attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein R3 and R4 are repeated a certain number of times and terminates with the carbohydrate mannose or sialic acid attached to the polymer. In certain instances, the present invention relates to the aforementioned polymer which includes a polymer or copolymer of a polyester, polyamide, polyether, or polycarbonate at the center or periphery of the polymers above taken from the structures below.
Structure I
R7 R9 R11 R13 J
-h+t R ~8-"fcrH R R1100 RR-11K R R-1122>4 R-14"-|r Structure II
Structure III In certain instances, the present invention relates to the aforementioned polymer block or random copolymer which includes at least one terminal or internal crosslinkable group selected from the group consisting of amines, thiols, amides, phosphates, sulphates, hydroxides, alkenes, and alkynes.
In certain instances, the present invention relates to the aforementioned polymer wherein X, Y, M is O, S, N-H, N-R, wherein R is -H, CH2, CR2 or a chain as defined above, Se or any isoelectronic species of oxygen.
In certain instances, the present invention relates to the aforementioned polymer wherein an amino acid(s) is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein a polypeptide(s) is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein an antibody(ies) or single chain antibody(ies) is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein a nucleotide(s) is attached to Z, A, R3, and/or R4. In certain instances, the present invention relates to the aforementioned polymer wherein a nucleoside(s) is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein an oligonucleotide(s) is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein a ligand(s) is attached to Z, A, R3, and/or R4 that binds to a biological receptor.
In certain instances, the present invention relates to the aforementioned polymer wherein a pharmaceutical agent(s) is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein a carbohydrate(s) is attached to Z, A, R3, and/or R4. In certain instances, the present invention relates to the aforementioned polymer wherein a PET or MRI contrast agent(s) is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein the contrast agent is Gd(DPTA).
In certain instances, the present invention relates to the aforementioned polymer wherein an iodated compound(s) for X-ray imagaging is attached to Z, A, R3, and/or R4.
In certain instances, the present invention relates to the aforementioned polymer wherein a pharmaceutical agent(s) is attached to Z, A, R3, and/or R4 and is at least one selected from the group consisting of antibacterial, anticancer, anti-inflammatory, and antiviral. In certain instances, the present invention relates to the aforementioned polymer wherein the carbohydrate is mannose or sialic acid is covalently attached to the polymer.
Another aspect of the present invention relates to a surgical procedure which comprises using a photopolymerizable, or chemically crosslinkable, or non-covalently crosslinkable dendritic polymer or copolymer. Another aspect of the present invention relates to an ophthalimic surgical procedure wherein said dendritic polymer or copolymer is dissolved or suspended in an non-aqueous liquid such as soybean oil, mineral oil, corn oil, rapeseed oil, coconut oil, olive oil, saflower oil, cottonseed oil, aliphatic, cycloaliphatic or aromatic hydrocarbons having 4-30 carbon atoms, aliphatic or aromatic alcohols having 1-30 carbon atoms, aliphatic or aromatic esters having 2-30 carbon atoms, alkyl, aryl or cyclic ethers having 2-30 carbon atoms, alkyl or aryl halides having 1-30 carbon atoms and optionally having more than one halogen substituent, ketones having 3-30 carbon atoms, polyalkylene glycol or combinations of any two or more thereof.
In certain instances, the present invention relates to the ophthalimic surgical procedure wherein the supramolecular structure of the dendrimer is an emulsion.
In certain instances, the present invention relates to the dendritic polymer or copolymer which optionally contains at least one stereochemical center. In certain instances, the present invention relates to the dendritic polymer or copolymer which is of D or L configuration.
In certain instances, the present invention relates to the dendritic polymer or copolymer wherein the final dendritic polymer or monomer is chiral or is achiral.
In certain instances, the present invention relates to the dendritic polymer or copolymer which contains at least one site where the branching is incomplete.
In certain instances, the present invention relates to a crosslinkable/photocrosslinkable/reactionary dendritic polymer or copolymer which contains at least one site where the branching is incomplete.
In certain instances, the present invention relates to a crosslinkable/photocrosslinkable/reactionary dendritic polymer or copolymer which contains at least one site where the branching is incomplete which forms a hydrogel.
In certain instances, the present invention relates to a crosslinkable/photocrosslinkable/reactionary dendritic polymer or copolymer which contains at least one site where the branching is incomplete and used for drug delivery. In certain instances, the present invention relates to a crosslinkable/photocrosslinkable/reactionary dendritic polymer or copolymer which contains at least one site where the branching is incomplete and used as a lens.
In certain instances, the present invention relates to a dendritic polymer or copolymer made by a convergent or divergent synthesis.
In certain instances, the dendritic polymer of the invention relates to
Sterilization Procedures
A variety of procedures are known in the art for sterilizing a chemical composition. Sterilization may be accomplished by chemical, physical, or irradiation techniques.
Examples of chemical methods include exposure to ethylene oxide or hydrogen peroxide vapor. Examples of physical methods include sterilization by heat (dry or moist), retort canning, and filtration. The British Pharmacopoeia recommends heating at a minimum of 160 0C for not less than 2 hours, a minimum of 170 0C for not less than 1 hour and a minimum of 180 0C for not less than 30 minutes for effective sterilization. For examples of heat sterilization, see U.S. Patent 6,136,326, which is hereby incorporated by reference. Passing the chemical composition through a membrane can be used to sterilize a composition. For example, the composition is filtered through a small pore filter such as a
0.22 micron filter which comprises material inert to the composition being filtered. In certain instances, the filtration is conducted in a Class 100,000 or better clean room. Examples of irradiation methods include gamma irradiation, electron beam irradiation, microwave irradiation, and irradiation using visible light. One preferred method is electron beam irradiation, as described in U.S. Patents 6,743,858; 6,248,800; and 6,143,805, each of which is hereby incorporated by reference.
There are several sources for electron beam irradiation. The two main groups of electron beam accelerators are: (1) a Dynamitron, which uses an insulated core transformer, and (2) radio frequency (RF) linear accelerators (linacs). The Dynamitron is a particle accelerator (4.5 MeV) designed to impart energy to electrons. The high energy electrons are generated and accelerated by the electrostatic fields of the accelerator electrodes arranged within the length of the glass-insulated beam tube (acceleration tube). These electrons, traveling through an extension of the evacuation beam tube and beam transport (drift pipe) are subjected to a magnet deflection system in order to produce a "scanned" beam, prior to leaving the vacuum enclosure through a beam window. The dose can be adjusted with the control of the percent scan, the beam current, and the conveyor speed. In certain instances, the electron-beam radiation employed may be maintained at an initial fluence of at least about 2 μCurie/cm2, at least about 5 μCurie/cm2, at least about 8 μCurie/cm2, or at least about 10 μCurie/cm2. In certain instances, the electron-beam radiation employed has an initial fluence of from about 2 to about 25 μCurie/cm2. In certain instances, the electron- beam dosage is from about 5 to 50 kGray, or from about 15 to about 20 kGray with the specific dosage being selected relative to the density of material being subjected to electron-beam radiation as well as the amount of bioburden estimated to be therein. Such factors are well within the skill of the art. The composition to be sterilized may be in any type of at least partially electron beam permeable container such as glass or plastic. In embodiments of the present invention, the container may be sealed or have an opening. Examples of glass containers include ampules, vials, syringes, pipettes, applicators, and the like. The penetration of electron beam irradiation is a function of the packaging. If there is not enough penetration from the side of a stationary electron beam, the container may be flipped or rotated to achieve adequate penetration. Alternatively, the electron beam source can be moved about a
stationary package. In order to determine the dose distribution and dose penetration in product load, a dose map can be performed. This will identify the minimum and maximum dose zone within a product.
Procedures for sterilization using visible light are described in U.S. Patent 6,579,916, which is hereby incorporated by reference. The visible light for sterilization can be generated using any conventional generator of sufficient power and breadth of wavelength to effect sterilization. Generators are commercially available under the tradename PureBright® in-line sterilization systems from PurePulse Technologies, Inc. 4241 Ponderosa Ave, San Diego, Calif. 92123, USA. The PureBright® in-line sterilization system employs visible light to sterilize clear liquids at an intensity approximately 90000 times greater than surface sunlight. If the amount of UV light penetration is of concern, conventional UV absorbing materials can be used to filter out the UV light.
In a preferred embodiment, the composition is sterilized to provide a Sterility Assurance Level (SAL) of at least about 10"3. The Sterility Assurance Level measurement standard is described, for example, in ISO/CD 14937, the entire disclosure of which is incorporated herein by reference. In certain embodiments, the Sterility Assurance Level may be at least about 10"4, at least about 10"5, or at least about 10"6.
Delivery Systems The materials used to repair the cartilaginous tissue may be delivered to the cartilage defect of a patient using a large number of known delivery devices. For example, the delivery system may be a single-barrel syringe system. In certain instances, the single- barrel syringe is a double acting, single-barrel syringe system as displayed in Figure 10. In certain situations, a double- or multi-barrel syringe system, as displayed in Figure 11 , may be preferable. In instances where the polymerizable dendrimer is mixed with a polymerization agent prior to delivering the solution to the cartilage defect of a patient, a delivery device that flows two or more streams of liquid in a mixing chamber may be preferable. Alternatively, a delivery device that mixes two solids and two liquids and then separately flows these streams of liquid to a mixing chamber may be advantageous. In certain instances, a delivery system is used to deliver materials to the cartilage defect of a patient, wherein at least two dry, reactive components are stored together in a dry state and
introduced into a liquid component(s) at the time of use to form a mixture that forms a hydrogel.
In certain instances, it may be advantageous the mix the components used to form the hydrogel by a tortuous path mixing element. For example, the two components could be mixed (without gelation) prior to applying the mixture to a patient. The pH of the mixing solution may be adjusted in order to slow or prevent crosslinking of hydrogel components. Once the components used to form the hydrogel are mixed, the resultant solution may be contacted with a frit or resin designed to raise or lower the pH to a level suitable for crosslinking. For example, PEG-SPA and Lys3Cys4 could be mixed during packaging and dissolved prior to use in a buffer designed to provide a solution with a pH of about 6. The solution is mixed, and then the solution is contacted with a resin embedded in the delivery device. The resin would raise the pH to about 7 for initiate crosslinking.
Methods of the Invention
One aspect of the present invention relates to a method of repairing cartilaginous tissue, comprising the steps of: applying an effective amount of a dendrimeric compound of formula Ia or formula Ib to a cartilage defect of a patient and exposing said dendrimeric compound to a polymerization agent sufficient to polymerize said dendrimeric compound, wherein said polymerization agent is ultraviolet light, visible light, a compound of formula II, a compound of formula III, a compound of formula IV, a compound of formula V, or an oxidizing agent, wherein formula Ia is represented by:
A1— X1-B— X1— A2
Ia wherein
A is alkyl, aryl, aralkyl, -Si(R
J 3)
X3,
or
A3 represents independently for each occurrence alkyl, cycloalkyl, heteroalkyl, heterocycloalkyl, aryl, heteroaryl, or aralkyl;
Y
1 represents independently for each occurrence R
4, A
4,
Z1 represents independently for each occurrence -X1 -R4 , E, or
Y
2 represents independently for each occurrence R
5, A
4,
Z represents independently for each occurrence -X -R , E, or
Y
3 represents independently for each occurrence R
6, A
4,
Z3 represents independently for each occurrence -X1 -R6 , E, or
-γ1-
Z
4 represents independently for each occurrence -X
1 -R
7, E, or
Y
5 represents independently for each occurrence R
8, A
4,
Z represents independently for each occurrence -X 1 - τR,8 , E, or
Y
6 represents independently for each occurrence R
9, A
4,
R represents independently for each occurrence H, alkyl, or halogen;
R represents independently for each occurrence H, alkyl, -OH, -N(R )2, -SH, hydroxyalkyl, or -[C(R')2]dR16;
R represents independently for each occurrence alkyl, aryl, or aralkyl; R4, R5, R6, R7, R8, and R9 are H;
R10 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R1 ' represents independently for each occurrence H, -OH, -N(R10)2, -SH, alkyl, hydroxyalkyl, or -[C(R')2]dR16;
R12 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R13 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R14 represents independently for each occurrence H, alkyl, or -CO2R10; R represents independently for each occurrence H, alkyl, or -OR ; R16 represents independently for each occurrence phenyl, hydroxyphenyl, pyrrolidyl, imidazolyl, indolyl, -N(R10)2, -SH, -S-alkyl, -CO2R10, -C(O)N(R10)2, or - C(NH2)N(R10)2;
d represents independently for each occurrence 1, 2, 3, 4, 5, or 6; n represents independently for each occurrence 1, 2, 3, 4, 5, or 6; p1 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p2 represents independently for each occurrence 0, 1, 2, 3, or 4; p3 represents independently for each occurrence 1, 2, or 3;
p represents independently for each occurrence 0, 1, 2, or 3; t represents independently for each occurrence 2, 3, 4, or 5 in accord with the rules of valence;
v1 and v2 each represent independently for each occurrence 2, 3, or 4; w1 and w2 each represent independently for each occurrence an integer from about 5 to about 1000, inclusive; x is 1, 2, or 3; y is O, 1, 2, 3, 4, or 5; z1 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; z2 and z3 each represent independently for each occurrence 1, 2, 3, 4, or 5;
X1 and X2 each represent independently for each occurrence O or -N(R10)-; X3 represents independently for each occurrence O, N(R10), or C(R15)(CO2R10);
H2N-CH-C- 1 CH2 CH2 CH2
O NH
H2N-CH-C-^ C=NH
A4 represents independently for each occurrence CH3 , NH2 )
H2N-
E represents independently for each occurrence
; and provided that R only occurs once, R only occurs once, R only occurs once, R only occurs once, R only occurs once, and R
9 only occurs once; said formula Ib is represented by:
Ib or a pharmaceutically acceptable salt, solvate, or hydrate thereof, wherein
X represents independently for each occurrence O or -N(R 22N )-;
R17 represents independently for each occurrence H, -(C(R19)2)hSH, -
R19 N~R18
C(O)(C(R19)2)hSH, -CO2(C(R19)2)hSH, -C(O)N(R18)(C(R19)2)hSH, R18
R18 represents independently for each occurrence H or alkyl; R19 represents independently for each occurrence H, halogen, or alkyl;
R20 represents independently for each occurrence H or alkyl; R21 represents independently for each occurrence H, -(C(RI9)2)hSH, -
C(O)(C(R
19)
2)
hSH, -CO
2(C(R
19)
2)
hSH, -C(O)N(R
18)(C(R
19)
2)
hSH,
or
R
22 represents independently for each occurrence H, alkyl, aryl, or aralkyl; n
1 and h each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p
5 represents independently for each occurrence 1, 2, 3, 4, or 5; v represents independently for each occurrence 2, 3, or 4; and w is an integer in the range of about 5 to about 1000, inclusive; said formula II is represented by:
II wherein
R
1"11 represents independently for each occurrence H or
R2"11 represents independently for each occurrence H or alkyl; R3"11 represents independently for each occurrence H, halogen, or alkyl; R4"11 represents independently for each occurrence alkyl, aryl, or aralkyl;
R
" represents independently for each occurrence H or
and z represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; said formula HI is represented by:
R-I-IIl Q-I-IIl R-I-III
III wherein
,3-HI
R1-1" is -(C(R2-m)2)xC(O)H, -C(O)(C(Rz-'")2)yC(O)H, -(C(R""^
C(O)(C(R2-11I)2)yC(O)R3-"1, -(C(R2-m)2)xR4-Iπ, -C(O)(C(R2"111)^4-111, or
R > 2- "III represents independently for each occurrence H, alkyl, or halogen;
R ,3- "m represents independently for each occurrence fluoroalkyl, chloroalkyl, -
R " represents independently for each occurrence -N=C=O, -N=C=S,
R5-IIl
N -R5"111 \ / \ -R5-III
R'5-lll R5-lll ^ or R7S-HI V-III .
R5"1" represents independently for each occurrence H, alkyl, or aralkyl;
B , 1-
'HI is alkyl diradical, heteroalkyl diradical, or
x represents independently for each occurrence 0, 1, 2, 3, 4, 5, 6, 7, or 8;
y represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8;
v represents independently for each occurrence 2, 3, or 4; and
w is an integer in the range of about 5 to about 1000, inclusive;
said formula IV is represented by:
A1— X1-B— X1— A2
IV wherein
A i' , i-βs V
A
2 is alkyl, aryl, aralkyl,r -Si(R
3)
3,
or
A3 represents independently for each occurrence alkyl, cycloalkyl, heteroalkyl, heterocycloalkyl, aryl, heteroaryl, or aralkyl;
esents independently for each occurrence R
4,
R /p2 R
1
Z represents independently for each occurrence -X ^l - rR.4 , E, or
Y
2 represents independently for each occurrence R
5,
Z2 represents independently for each occurrence -X'-R5, E, or hχ1-(?)-f χ2"γ3) t.
Z represents independently for each occurrence -X -R , E, or
h
χ1-@)-f
χ2-
γ4)
t.
Y
4 represents independently for each occurrence R
7,
Z represents independently for each occurrence -X 1 - rR>7 , E, or
h*1-@-(~χ2~γ5) t.
Y represents independently for each occurrence R ,
Z5 represents independently for each occurrence -X1 -R , E, or
Y represents independently for each occurrence
R represents independently for each occurrence H, alkyl, or halogen; l(k
R represents independently for each occurrence H, alkyl, -OH, -N(R )2, -SH, hydroxyalkyl, or -[C(R')2]dR16;
R3 represents independently for each occurrence alkyl, aryl, or aralkyl;
R4, R5, R6, R7, R8, and R9 are H;
R10 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R1 ' represents independently for each occurrence H, -OH, -N(R10)2, -SH, alkyl, hydroxyalkyl, or -[C(R1),],^16;
R12 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R13 represents independently for each occurrence H, alkyl, aryl, or aralkyl;
R14 represents independently for each occurrence H, alkyl, or -CO2R10; R15 represents independently for each occurrence H, alkyl, or -OR10;
R16 represents independently for each occurrence phenyl, hydroxyphenyl, pyrrolidyl, imidazolyl, indolyl, -N(R10)2, -SH, -S-alkyl, -CO2R10, -C(O)N(R10)2, or - C(NH2)N(R10)2;
n represents independently for each occurrence 1, 2, 3, 4, 5, or 6; p represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 0, 1, 2, 3, or 4; p3 represents independently for each occurrence 1, 2, or 3; p4 represents independently for each occurrence 0, 1, 2, or 3; d represents independently for each occurrence 1, 2, 3, 4, 5, or 6; t represents independently for each occurrence 2, 3, 4, or 5 in accord with the rules of valence;
v1 and v2 each represent independently for each occurrence 2, 3, or 4; w1 and w2 each represent independently for each occurrence an integer from about 5 to about 1000, inclusive; x is 1, 2, or 3; y is O, 1, 2, 3, 4, or 5; z1 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; z2 and z3 each represent independently for each occurrence 1 , 2, 3, 4, or 5;
X1 and X2 each represent independently for each occurrence O or -N(R10)-;
X3 represents independently for each occurrence O, N(R10), or C(R15XCO2R1 °); E represents independently for each occurrence H, -[C(R1^]nC(O)H, -(C(R13)2)XR17,
R17 represents independently for each occurrence -N=C=O, -N=C=S, R13 R13 ,
^R13 N
_R13 or ^ R13 R13 ;and
said formula V is represented by:
or a pharmaceutically acceptable salt, solvate, or hydrate thereof, wherein
X6 represents independently for each occurrence O or -N(R30)-; R23 represents independently for each occurrence
R ,24 represents independently for each occurrence H or alkyl;
R ,25 represents independently for each occurrence H, halogen, or alkyl;
R ,26 represents independently for each occurrence H or alkyl;
R ,27 represents independently for each occurrence H, alkyl, or halogen;
R28 represents independently for each occurrence H, alkyl, -OH, -N(R30)2, -SH, or hydroxyalkyl;
R ,29 represents independently for each occurrence H, -OH, -N(R 3O )x2, -SH, alkyl, or hydroxyalkyl;
R30 and R31 represent independently for each occurrence H, alkyl, aryl, or aralkyl; Z6 represents independently for each occurrence E1 or
Z7 represents independently for each occurrence E1 or
R
33 represents independently for each occurrence
R
34 represents independently for each occurrence H, alkyl, or -CO
2R
30; E
1 represents independently for each occurrence H, -[C(R
24)
2]
jC(O)H, -
O
-R 13
R35 represents independently for each occurrence -N=C=O, -N=C=S, R13 R13 , .R13
,R13 or ' R13 R13 ; p6 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p7 represents independently for each occurrence 0, 1, 2, 3, or 4; p8 represents independently for each occurrence 1, 2, or 3; p9 represents independently for each occurrence 0, 1, 2, or 3; n2 and j each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; m1 represents independently for each occurrence 1 or 2; v represents independently for each occurrence 2, 3, or 4; and w is an integer in the range of about 5 to about 1000, inclusive.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula Ia, and said polymerization agent is ultraviolet light, visible light, a compound of formula II, a compound of formula III, or an oxidizing agent.
In certain instances, the present invention relates to the aforementioned method,
wherein A is
, and m is 1 or 2.
In certain instances, the present invention relates to the aforementioned method,
, or -Si(R )
3; wherein, m is 1 or 2.
In certain instances, the present invention relates to the aforementioned method, wherein Z1 represents independently for each occurrence -X'-R4 or
In certain instances, the present invention relates to the aforementioned method, wherein Z2 represents independently for each occurrence -X'-R5 or
In certain instances, the present invention relates to the aforementioned method, wherein Z represents independently for each occurrence -X 1 - rR>6 or
In certain instances, the present invention relates to the aforementioned method, wherein Z4 represents independently for each occurrence -X'-R7 or
2.
In certain instances, the present invention relates to the aforementioned method, wherein Z
5 represents independently for each occurrence -X
1 -R
8 or
In certain instances, the present invention relates to the aforementioned method, wherein X1 is O.
In certain instances, the present invention relates to the aforementioned method, wherein X and X are O.
In certain instances, the present invention relates to the aforementioned method, wherein n is 1. In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, 3, or 4.
In certain instances, the present invention relates to the aforementioned method, wherein p2 is 1.
In certain instances, the present invention relates to the aforementioned method, wherein R1 is H.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
X ' P , A
2 is
, m
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
and said polymerization agent is a compound of formula III.
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, A
2 , m
In certain instances, the present invention relates to the aforementioned method,
of
In certain instances, the present invention relates to the aforementioned method,
wherein R' is H, B is
A Λ
2Ms
m
the
Y
4 groups are
and said polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, A
2 m
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 1, 2, 3, or 4.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 4. In certain instances, the present invention relates to the aforementioned method, wherein m is 1.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
1
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R is H, B is
, m is 1
In certain instances, the present invention relates to the aforementioned method,
wherein R is H, B is
, m is 1
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
x 7 P , A
2 is , m is 1
and said polymerization agent is compound of formula III.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
X ' P , A
2 , m is 1
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 1, 2, 3, or 4.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 4.
In certain instances, the present invention relates to the aforementioned method, wherein m is 1. In certain instances, the present invention relates to the aforementioned method, wherein R2 is (Ci-C3)alkyl.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is VR
1 RV pI VR
1 RVv
I
, and v
1 is
2.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
v
1 is 2, A
2
In certain instances, the present invention relates to the aforementioned method,
polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R is H, B is
V
1 is 2, A
2
In certain instances, the present invention relates to the aforementioned method,
polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method, wherin w
1 is an integer in the range of about 50 to about 250.
In certain instances, the present invention relates to the aforementioned method, wherein w1 is an integer in the range of about 60 to about 90.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein m is 1.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, and R3 is (Ci-C5)alkyl.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, R3 is (Ci-C5)alkyl, and w1 is an integer in the range of about 60 to about 90.
In certain instances, the present invention relates to the aforementioned method,
i
In certain instances, the present invention relates to the aforementioned method,
wherein R is H, B is
A
2 is
, R
3 is alkyl, v
2 is
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, A
2 is
, R
3 is alkyl, v
2 is
and said polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, A
2 is
, R
3 is alkyl, v
2 is
In certain instances, the present invention relates to the aforementioned method,
i iS
In certain instances, the present invention relates to the aforementioned method,
and said polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method, wherein p
1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein m is 1.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, and R3 is (Ci-C5)alkyl.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, and R3 is (Ci-C5)alkyl, and w2 is an integer in the range of about 60 to about 90.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
is 1, or 2, Y
1 is
x V , Z
1
, Y
2 is
In certain instances, the present invention relates to the aforementioned method,
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula II. In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1'"1 is -CH2C(O)H, and R2-1" is H. In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1"111 is -CH2C(O)H, R "'"
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R l- "III is -CH2C(O)H, R J 2-III
is H, B
1-"
1 is
and w is an integer in the range of about 60-
90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R l-
"III is -(C(R
*
")
2)
XC(O)R
J-"' or -
C(O)(C(R^
ul)
2)
yC(O)R
>IU, R'-"
1 is H, and R
MU or
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1"111 is -(C(R2'
)
2)χC(O)R
J""' or -C(O)(C
■ TT
s H, r R, 3'-
"l"ll
1 is or
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, RMπ is -(C(R2"
in , 3-IH ,2-IIN
)
2)
XC(O)R'
"'" or -C(O)(C(R
z'm)
2)
yC(O)R , 3
>-I
mH, r R» 2
z-
"i
mπ i
•s H, R
^" is or
B
1-
111 is
-
w , and w is an integer in the range of about 60-90.
In certain instances, the present invention relates to the aforementioned method, wherein said compound of formula Ia is
n is an integer in the range of about 70 to about 80, and said polymerization agent is UV light.
In certain embodiments, the present invention relates to the aforementioned method, wherein said dendrimeric compound is compound of formula Ib.
In certain embodiments, the present invention relates to the aforementioned method, wherein v is 2.
In certain embodiments, the present invention relates to the aforementioned method, wherein X5 is -N(H)-. In certain embodiments, the present invention relates to the aforementioned method, wherein R18 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R19 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R20 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 20-500.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 40-250. In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 60-90.
In certain embodiments, the present invention relates to the aforementioned method, said compound of formula Ib is
In certain embodiments, the present invention relates to the aforementioned method, said polymerization agent is a compound of formula V.
In certain embodiments, the present invention relates to the aforementioned method, wherein v is 2. In certain embodiments, the present invention relates to the aforementioned method, wherein X6 is -N(H)-.
In certain embodiments, the present invention relates to the aforementioned method, wherein R24 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R25 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R26 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 20-500. In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 40-250.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 60-90.
In certain embodiments, the present invention relates to the aforementioned method, wherein R represents independently for each occurrence
In certain embodiments, the present invention relates to the aforementioned method, wherein R23 represents independently for each occurrence
In certain embodiments, the present invention relates to the aforementioned method, said compound of formula V is
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is an oxidizing agent.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is O2.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is ultraviolet light or visible light. In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is ultraviolet light.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is light with a λ of 400-600 ran.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is light with a λ of 450-550 nm.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is light with a λ of 488-514 nm.
In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a primate, bovine, equine, feline, or canine. In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a human.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear, strain, void, fibrillation, or a decrease in the amount of cartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear.
In certain embodiments, the present invention relates to the aforementioned method, wherein said tear is less than about 5 mm long.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in articular cartilage. In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in fϊbrocartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in the meniscus.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 10 cm2 in size.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 5 cm2 in size.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 1 cm2 in size. In certain embodiments, the present invention relates to the aforementioned method, wherein said compound of formula Ia is dissolved in at least one solvent, and said compound of formula Ia has a concentration in the range of about 2% w/w to about 40% w/w.
In certain embodiments, the present invention relates to the aforementioned method, wherein said compound of formula Ia is dissolved in at least one solvent, and said compound of formula Ia has a concentration in the range of about 5% w/w to about 20% w/w.
In certain embodiments, the present invention relates to the aforementioned method, wherein said compound of formula Ia is dissolved in at least one solvent, and said compound of formula Ia has a concentration in the range of about 6% w/w to about 10% w/w. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing a photoinitiator with said compound of formula Ia prior to exposing said compound of formula Ia to said polymerization agent.
In certain embodiments, the present invention relates to the aforementioned method, wherein said photoinitiator is eosin-Y. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing a natural polymer with said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, wherein said natural polymer is HA, collagen, or a GAG fragment. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing at least one cell with said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cell is a cartilage cell or a stem cell.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent, wherein said polymerization agent is selected from the group consisting of a compound of formula II, a compound of formula III, a compound of formula IV, and a compound of formula V.
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is performed by treatment with ethylene oxide, hydrogen peroxide, heat, gamma irradiation, electron beam irradiation, microwave irradiation, or visible light irradiation. In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about 10"
3
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about 10" 6.
Another aspect of the present invention relates to a method of repairing cartilaginous tissue, comprising the steps of: applying an effective amount of a dendrimeric compound of formulae VI , VII, VIII, or IX to a cartilage defect of a patient and exposing said dendrimeric compound to a polymerization agent sufficient to polymerize said dendrimeric compound, wherein said polymerization agent is an oxidizing agent or a compound of formula X, wherein formula VI is represented by:
VI or a pharmaceutically acceptable salt, solvate, or hydrate thereof, wherein
R1 represents independently for each occurrence H, OH, -(C(R3)2)mN(R2)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
R2 represents independently for each occurrence H or alkyl; R3 represents independently for each occurrence H, halogen, or alkyl; R4 represents independently for each occurrence alkyl, aryl, or aralkyl;
R5 represents independently for each occurrence OH, -(C(R3)2)mN(R2)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; and p is 1, 2, 3, 4, or 5; formula VII is represented by:
VII wherein
R
1 represents independently for each occurrence H, -(C(R
3)
2)
mN(H)R
4, - (C(R
3)
2)
mN(R
4)OH, -(C(R
3)
2)
mSH, -C(O)(C(R
3)
2)
mSH, -CO
2(C(R
3)
2)
mSH, -
C(O)N(R
2)(C(R
3)
2)
mSH,
R2 represents independently for each occurrence H, alkyl, or -(C(R3)2)XOR1 ; R3 represents independently for each occurrence H, halogen, or alkyl; R4 represents independently for each occurrence H, alkyl, aryl, or aralkyl;
R5 represents independently for each occurrence OH, -(C(R3)2)mN(R2)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 1, 2, 3, 4, or 5; x represents independently for each occurrence 1, 2, 3, or 4; and y is O, 1, 2, 3, or 4; formula VIII is represented by:
VIII wherein
R
1 represents independently for each occurrence H, -(C(R
3)2)
mSH, - o /R
3R
3\ P
R3 N~R4 C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH, R4
R2 represents independently for each occurrence H, alkyl, -(C(R3)2)mYR1, OH, - (C(R3)2)mN(H)R4, -(C(R3)2)mN(R4)OH, -(C(R3)2)mSH, -C(O)(C(R3)2)mSH, -
CO
2(C(R
3)
2)
mSH, -C(O)N(R
2)(C(R
3)
2)
mSH,
R3 represents independently for each occurrence H, halogen, or alkyl; R4 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R5 represents independently for each occurrence OH, -(C(R3)2)mN(R2)OH, -
(C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
Y represents independently for each occurrence O or NR
4; n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 1, 2, 3, 4, or 5; and x represents independently for each occurrence 1, 2, 3, or 4; formula IX is represented by:
IX wherein
R1 represents independently for each occurrence H, -(C(R )2)mN(H)R , (C(R3)2)mN(R4)OH, -(C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -
R represents independently for each occurrence alkyl, aryl, or aralkyl; R3 represents independently for each occurrence H, halogen, or alkyl; R represents independently for each occurrence H, alkyl, aryl, or aralkyl;
R5 represents independently for each occurrence OH, -(C(R3)2)mN(R4)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 1, 2, 3, 4, or 5; and x is 1 or 2; and formula X is represented by:
R1-X_B_R1-X
wherein
2-
R , i'-"xΛ represents independently for each occurrence -(C(R >2-X\ )2)XC(O)H, -C(O)(C(R' x)2)yC(0)H, -(C(R2-χ)2)χC(O)R3-χ, -C(O)(C(R2-χ)2)yC(O)R3-χ, -(C(R2-χ)2)xR4-χ, -
R2"x represents independently for each occurrence H, alkyl, or halogen;
R3"x represents independently for each occurrence alkyl, fluoroalkyl, chloroalkyl, -
O
_R5-X
R >4-X represents independently for each occurrence -N=C=O, -N=C=S, R5 5'-xX R D5-X
.R5-χ
N'
.R5-X or ' R5"x R5-χ ;
R , 5- 'X represents independently for each occurrence H, alkyl, or aralkyl;
B is alkyl diradical, heteroalkyl diradical, or
•
v2"x represents independently for each occurrence 2, 3, or 4; w2"x is an integer in the range of about 5 to 1000, inclusive; and x and y each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, 8, or 9.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is an oxidizing agent.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is O2. In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X.
In certain instances, the present invention relates to the aforementioned method, w2" x is an integer in the range of about 50 to about 250.
In certain instances, the present invention relates to the aforementioned method, w2' x is an integer in the range of about 60 to about 90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R!"x is -CH2C(O)H, and R2 X is H.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R'"X is -CH2C(O)H, R2'x is
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, Rux is -CH2C(O)H, R2 X is
H, B is
y
2-x j
s 2
( an(j
w 2-x j
s an j
nteger in the range of about 15-90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R*"x is -(C(R2"X)2)XC(O)R3"
or -C(O)(C(R'
"Λ)
2)
yC(O)R
J"Λ, R 2
Z-
"X
Λ i
■s H u, and i τ R>3
J-
"X
Λ is
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R'
"x is -(C(R
2"X)
2)
XC(O)R
3" or -C(O)(C(R'-
A)
2)
yC(O)
B is
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R!'x is -(C(R2 X)2)XC(O)R3"
is 2, and w
2"x is an integer in the range of about 15-90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, wherein B is
d
2"x, e
2"x, and g
2'x represent independently an integer greater than zero, provided that the sum of d
2'x, e
2"x, and g
2'x is an integer in the range of about 5 to 1000, inclusive.
In certain instances, the present invention relates to the aforementioned method, i- ,2-Xx ,3-X 2-Xx ,3-X D2-X ,3-X wherein, R , '"xΛ is -(C(R^Λ)2)XC(O)RJ-Λ or -C(O)(C(Rz-Λ)2)yC(O)R , R^Λ is H, and R>λ is
In certain instances, the present invention relates to the aforementioned method,
s is an integer in the range of about 1 -20, inclusive.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI.
In certain instances, the present invention relates to the aforementioned method, wherein n is 3, 4, or 5.
In certain instances, the present invention relates to the aforementioned method, wherein n is 4. In certain instances, the present invention relates to the aforementioned method, wherein R2 is H.
In certain instances, the present invention relates to the aforementioned method, wherein R3 is H.
In certain instances, the present invention relates to the aforementioned method, wherein R4 is alkyl.
In certain instances, the present invention relates to the aforementioned method, wherein R4 is methyl or ethyl.
In certain instances, the present invention relates to the aforementioned method, wherein n is 4, R2 and R3 is H, and R4 is alkyl. In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R is R SH R2 , and p is 1.
In certain instances, the present invention relates to the aforementioned method,
wherein n is 4, R
2 and R
3 are H, R
4 is methyl, R
1 is
R
2 , and p is 1.
In certain instances, the present invention relates to the aforementioned method,
wherein n is 4, R
2 and R
3 are H, R
4 is methyl, R
1 is
and p is 1.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and a Bronsted acid. In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and HA, wherein A is halogen or -O2CR6, and R6 is alkyl, fluoroalkyl, aryl, or aralkyl.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said
pharmaceutically acceptable salt is a complex formed by said compound of formula VI and an acid selected from group consisting of HCl and HBr.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and HO2CR6, wherein R6 is fluoroalkyl.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and CF3CO2H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VII.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VII, x and y are 1, R is - CH2OR1, and R3 is H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VII, x is 1 , y is O, and R2 and R3 are H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VIII.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VIII, x is 2, Y is O, R2 is - CH2CH2OR1, and R3 is H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VIII, x is 2, Y is NR4, and R2 and R3 are H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula IX.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula IX, R2 is methyl, and x is 2.
In certain instances, the present invention relates to the aforementioned method, further comprising the step of exposing said dendrimeric compound to a compound of formula XI, wherein formula XI is represented by:
R1-XI_BXI_R1-XI
XI
wherein
R'"XI represents independently for each occurrence -(C(R2'X1)2)XC(O)H, -C(O)(C(R2"
XH )2)yC(O)H, -(C(R ,2'--XΛK1)2)XC(O)R , 3J--XΛI1, -C(O)(C(R'-Λ1)2)yC(O)R>ΛI, -(C(R'-λl)2)χR* , -
R ,2- "XI represents independently for each occurrence H, alkyl, or halogen;
R3'XI represents independently for each occurrence alkyl, fluoroalkyl, chloroalkyl, -
R4'XI represents independently for each occurrence -N=C=O, -N=C=S,
R5-Xl
O N i A pSXI > / \ p5-XI
R5-xΛR5-xι , or R5-XI XR5-XI .
R5"XI represents independently for each occurrence H, alkyl, or aralkyl;
B iXI is alkyl diradical, heteroalkyl diradical, or
v 2-
"Xl represents independently for each occurrence 2, 3, or 4; w
2 XI is an integer in the range of about 5 to 1000, inclusive; and x and y each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, 8, or 9.
In certain embodiments, the present invention relates to the aforementioned method,
In certain embodiments, the present invention relates to the aforementioned method,
wherein, B XI is
w2-
χι t v2-x
i is 2) R'-
X1 is -(C(R
2-
χi)
2)
χC(0)R
30α In certain embodiments, the present invention relates to the aforementioned method,
wherein, B XΛI1 R'-XI is -(C(R2-XI)2)XC(O)R3"XI
or -C(O)(C(R
2-
χi)
2)
yC(O)R
3-
χi, R
20CI is H, R > 3
>-X
ΛI
1 . is or
said polymerization agent is a compound of formula X, B is
e
2"x, and g
20C represent independently an integer greater than zero, provided that the sum of d
2"x, e
2"x, and g
2"x is an integer in the range of about 5 to 1000, inclusive.
In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a primate, bovine, equine, feline, or canine.
In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a human.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear, strain, void, fibrillation, or a decrease in the amount of cartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear.
In certain embodiments, the present invention relates to the aforementioned method, wherein said tear is less than about 5 mm long. In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in articular cartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in fibrocartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in the meniscus.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 10 cm2 in size.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 5 cm2 in size. In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 1 cm in size.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing a natural polymer with said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, wherein said natural polymer is HA, collagen, or a GAG fragment.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing at least one cell with said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cell is a cartilage cell or a stem cell. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent, wherein said polymerization agent is a compound of formula X.
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is performed by treatment with ethylene oxide, hydrogen peroxide, heat, gamma irradiation, electron beam irradiation, microwave irradiation, or visible light irradiation.
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about 10"
3
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about 10"
6
Another aspect of the present invention relates to a method of repairing cartilaginous tissue, comprising the steps of: exposing a dendrimeric compound of formula Ia or formula Ib to a polymerization agent to form a repair agent and applying said repair agent to a cartilage defect of a patient, wherein said polymerization agent is ultraviolet light, visible light, a compound of formula II, a compound of formula III, a compound of formula IV, a compound of formula V, or an oxidizing agent, wherein formula Ia is represented by:
A1— X1-B— X1— A2
Ia wherein
A
2 is alkyl, aryl, aralkyl, -Si(R
3)
3,
or
A represents independently for each occurrence alkyl, cycloalkyl, heteroalkyl, heterocycloalkyl, aryl, heteroaryl, or aralkyl;
Y represents independently for each occurrence R , A ,
Z represents independently for each occurrence -X 1 - rR>4 , E, or
Y represents independently for each occurrence R , A ,
Z2 represents independently for each occurrence -X'-R5, E, or hx1"(^)-(-χ2-γ3) t.
Y represents independently for each occurrence R 6 , A
A 4 ,
Z represents independently for each occurrence -X 1 - rR.6 , E, or
Y
4 represents independently for each occurrence R
7, A
4,
Z4 represents independently for each occurrence -X1 -R7, E, or
Y represents independently for each occurrence R , A ,
Z
5 represents independently for each occurrence -X
1 -R
8, E, or
Y
6 represents independently for each occurrence R
9, A
4,
R
1 represents independently for each occurrence H, alkyl, or halogen; R
2 represents independently for each occurrence H, alkyl, -OH, -N(R
10)
2, -SH, hydroxyalkyl, or -[C(R')
2]dR
16;
R represents independently for each occurrence alkyl, aryl, or aralkyl; τ R)60, τ R)7', R8, and Ry are H;
R , 10 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R1 ' represents independently for each occurrence H, -OH, -N(R10)2, -SH, alkyl, hydroxyalkyl, or -[C(R')2]dR16;
R1 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R13 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R14 represents independently for each occurrence H, alkyl, or -CO2R10; R15 represents independently for each occurrence H, alkyl, or -OR10; R1 represents independently for each occurrence phenyl, hydroxyphenyl, pyrrolidyl, imidazolyl, indolyl, -N(R10)2, -SH, -S-alkyl, -CO2R10, -C(O)N(R10)2, or - C(NH2)N(R10)2;
d represents independently for each occurrence 1, 2, 3, 4, 5, or 6; n represents independently for each occurrence 1, 2, 3, 4, 5, or 6; p1 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p2 represents independently for each occurrence 0, 1, 2, 3, or 4; p3 represents independently for each occurrence 1, 2, or 3; p4 represents independently for each occurrence 0, 1, 2, or 3; t represents independently for each occurrence 2, 3, 4, or 5 in accord with the rules of valence;
v1 and v2 each represent independently for each occurrence 2, 3, or 4; w1 and w2 each represent independently for each occurrence an integer from about 5 to about 1000, inclusive; x is 1, 2, or 3; y is O, 1, 2, 3, 4, or 5; z represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; z and z each represent independently for each occurrence 1, 2, 3, 4, or 5; X and X each represent independently for each occurrence O or -N(R10)-; X3 represents independently for each occurrence O, N(R10), or C(R15)(CO2R10);
H2N-CH-C- 1 CH2 CH2 CH2
O NH
H2N-CH-C-^ C=NH
A represents independently for each occurrence CH3 NH2
H2N-
H2N-
H2N-
E represents independently for each
provided that R
4 only occurs once, R
5 only occurs once, R
6 only occurs once, R
7 only occurs once, R
8 only occurs once, and R
9 only occurs once; said formula Ib is represented by:
Ib or a pharmaceutically acceptable salt, solvate, or hydrate thereof, wherein
X5 represents independently for each occurrence O or -N(R22)-; R17 represents independently for each occurrence H, -(C(R19)2)hSH, -
R19 N^R18
C(O)(C(R19)2)hSH, -CO2(C(R19)2)hSH, -C(O)N(R18)(C(R19)2)hSH, R18
R18 represents independently for each occurrence H or alkyl; R19 represents independently for each occurrence H, halogen, or alkyl; R20 represents independently for each occurrence H or alkyl;
R21 represents independently for each occurrence H, -(C(R19)2)hSH, -
R19 N-R18 C(O)(C(R19)2)hSH, -CO2(C(R19)2)hSH, -C(O)N(R18)(C(R19)2)hSH, R18 , or
R22 represents independently for each occurrence H, alkyl, aryl, or aralkyl; n1 and h each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 1, 2, 3, 4, or 5; v represents independently for each occurrence 2, 3, or 4; and w is an integer in the range of about 5 to about 1000, inclusive;
said formula II is represented by:
II wherein
R
1'11 represents independently for each occurrence H
or R
5""
R2"11 represents independently for each occurrence H or alkyl;
R3'" represents independently for each occurrence H, halogen, or alkyl;
R4"11 represents independently for each occurrence alkyl, aryl, or aralkyl; and
R
5 11 represents independently for each occurrence H
or and z represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; said formula HI is represented by:
R1-lll £1-111 R1-lll
III wherein
,3-m
R1 "1 is -(C(R2-m)2)xC(O)H, -C(O)(C(R2-m)2)yC(O)H, -(C(R2-πi)2)xC(O)Rj m, C(O)(C(R2-iπ)2)yC(O)R3-m, -(C(R2-m)2)xR4-m, -C(O)(C(R2-m)2)yR4-m, or
R2'111 represents independently for each occurrence H, alkyl, or halogen;
R3"1" represents independently for each occurrence fluoroalkyl, chloroalkyl, -
R " represents independently for each occurrence -N=C=O, -N=C=S,
R5-IIl
N
RS-"1 R5"111 , Or ^IM RS-III ;
R5'111 represents independently for each occurrence H, alkyl, or aralkyl;
B , 1-
'HI is alkyl diradical, heteroalkyl diradical, or
x represents independently for each occurrence 0, 1, 2, 3, 4, 5, 6, 7, or 8;
y represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8;
v represents independently for each occurrence 2, 3, or 4; and
w is an integer in the range of about 5 to about 1000, inclusive;
said formula IV is represented by:
A1— X1-B— X1— A2
IV wherein
A represents independently for each occurrence alkyl, cycloalkyl, heteroalkyl, heterocycloalkyl, aryl, heteroaryl, or aralkyl;
Z represents independently for each occurrence -X rl - rR.4 , E, or
Z2 represents independently for each occurrence -X1 -R5, E, or
Y
3 represents independently for each occurrence R
6,
Z represents independently for each occurrence -X 1 -R Ti 6 , E, or
Z represents independently for each occurrence -X 1 - τR.7 , E, or
Z represents independently for each occurrence -X -R , E, or
R represents independently for each occurrence H, alkyl, or halogen;
R2 represents independently for each occurrence H, alkyl, -OH, -N(R10)2, -SH, hydroxyalkyl, or -[C(R')2]dR16;
R3 represents independently for each occurrence alkyl, aryl, or aralkyl; R4, R5, R6, R7, R8, and R9 are H;
R10 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R1 ' represents independently for each occurrence H, -OH, -N(R10)2, -SH, alkyl, hydroxyalkyl, or -[C(R')2]dR16;
R12 represents independently for each occurrence H, alkyl, aryl, or aralkyl; R13 represents independently for each occurrence H, alkyl, aryl, or aralkyl;
R14 represents independently for each occurrence H, alkyl, or -CO2R10; R15 represents independently for each occurrence H, alkyl, or -OR10;
R16 represents independently for each occurrence phenyl, hydroxyphenyl, pyrrolidyl, imidazolyl, indolyl, -N(R10)2, -SH, -S-alkyl, -CO2R10, -C(O)N(R10)2, or - C(NH2)N(R10)2;
n represents independently for each occurrence 1, 2, 3, 4, 5, or 6; p1 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p2 represents independently for each occurrence 0, 1, 2, 3, or 4; p3 represents independently for each occurrence 1, 2, or 3; p4 represents independently for each occurrence 0, 1, 2, or 3; d represents independently for each occurrence 1, 2, 3, 4, 5, or 6; t represents independently for each occurrence 2, 3, 4, or 5 in accord with the rules of valence;
v1 and v2 each represent independently for each occurrence 2, 3, or 4; w1 and w2 each represent independently for each occurrence an integer from about 5 to about 1000, inclusive; x is 1, 2, or 3; y is O, 1, 2, 3, 4, or 5; z1 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; z2 and z3 each represent independently for each occurrence 1 , 2, 3, 4, or 5;
X1 and X2 each represent independently for each occurrence O or -N(R10)-;
X3 represents independently for each occurrence O, N(R10), or C(R15)(CO2R10); E represents independently for each occurrence H, -[C(R1^]nC(O)H, -(C(R13)2)XR17,
R represents independently for each occurrence -N=C=O, -N=C=S,
,R13
N'
-R13 or R13 R13 ; and
said formula V is represented by:
or a pharmaceutically acceptable salt, solvate, or hydrate thereof, wherein
X6 represents independently for each occurrence O or -N(R °)-; R23 represents independently for each occurrence
R »24 represents independently for each occurrence H or alkyl;
R25 represents independently for each occurrence H, halogen, or alkyl;
R represents independently for each occurrence H or alkyl;
R27 represents independently for each occurrence H, alkyl, or halogen;
R28 represents independently for each occurrence H, alkyl, -OH, -N(R30)2, -SH, or hydroxyalkyl;
R >29 represents independently for each occurrence H, -OH, -N(R 30x )2, -SH, alkyl, or hydroxyalkyl;
R30 and R31 represent independently for each occurrence H, alkyl, aryl, or aralkyl; Z6 represents independently for each occurrence E1 or
R ,32 represents independently for each occurrence
Z7 represents independently for each occurrence E1 or
R >33 represents independently for each occurrence
R
34 represents independently for each occurrence H, alkyl, or -CO
2R
30;
E1 represents independently for each occurrence H, -[C(R24)2]jC(O)H, -
O
E / \ ,p13
R35 represents independently for each occurrence -N=C=O, -N=C=S, R13 R13 ,
p
6 represents independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p
7 represents independently for each occurrence 0, 1, 2, 3, or 4; p
8 represents independently for each occurrence 1, 2, or 3; p
9 represents independently for each occurrence 0, 1, 2, or 3; n
2 and j each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; m
1 represents independently for each occurrence 1 or 2; v represents independently for each occurrence 2, 3, or 4; and w is an integer in the range of about 5 to about 1000, inclusive.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula Ia, and said polymerization agent is ultraviolet light, visible light, a compound of formula II, a compound of formula III, or an oxidizing agent.
In certain instances, the present invention relates to the aforementioned method,
wherein A
1
, and m is 1 or 2.
In certain instances, the present invention relates to the aforementioned method,
, or -Si(R
3)
3; wherein, m is 1 or 2.
In certain instances, the present invention relates to the aforementioned method, wherein Z1 represents independently for each occurrence -X1 -R4 or
In certain instances, the present invention relates to the aforementioned method, wherein Z2 represents independently for each occurrence -X1 -R5 or
2. In certain instances, the present invention relates to the aforementioned method, wherein Z represents independently for each occurrence -X '-R
6 or
In certain instances, the present invention relates to the aforementioned method, wherein Z4 represents independently for each occurrence -X1 -R7 or
2.
In certain instances, the present invention relates to the aforementioned method, wherein Z
5 represents independently for each occurrence -X
1 -R
8 or
In certain instances, the present invention relates to the aforementioned method, wherein X1 is O.
In certain instances, the present invention relates to the aforementioned method, wherein X1 and X2 are O.
In certain instances, the present invention relates to the aforementioned method, wherein n is 1. In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, 3, or 4.
In certain instances, the present invention relates to the aforementioned method, wherein p2 is 1.
In certain instances, the present invention relates to the aforementioned method, wherein R1 is H.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
is ultraviolet light or light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
and said polymerization agent is a compound of formula III.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, m
, and at least about 1/2 of
In certain instances, the present invention relates to the aforementioned method,
, at least about 1/2 of the
Y
4 groups are
and said polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
1 , A
2 is , m
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 1 , 2, 3, or 4.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 4. In certain instances, the present invention relates to the aforementioned method, wherein m is 1.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
, m is 1
In certain instances, the present invention relates to the aforementioned method,
wherein R is H, B is
, m is 1
In certain instances, the present invention relates to the aforementioned method,
wherein R is H, B is
, m is 1
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, A
2 is , m is 1
and said polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
A
2 , m is 1
In certain instances, the present invention relates to the aforementioned method,
and said polymerization agent is a compound of formula III.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
X 7 P , A
2 , m is 1
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 1, 2, 3, or 4.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 4.
In certain instances, the present invention relates to the aforementioned method, wherein m is 1. In certain instances, the present invention relates to the aforementioned method, wherein R2 is (Ci-C3)alkyl.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, and v is 2.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, v' is 2, A
2 In certain instances, the present invention relates to the aforementioned method,
polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, v
] is 2, A
2
i :s,
In certain instances, the present invention relates to the aforementioned method,
polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method, wherein w
1 is an integer in the range of about 50 to about 250.
In certain instances, the present invention relates to the aforementioned method, wherein w1 is an integer in the range of about 60 to about 90.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein m is 1.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, and R3 is (Ci-C5)alkyl.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, R3 is (Ci-C5)alkyl, and w1 is an integer in the range of about 60 to about 90.
In certain instances, the present invention relates to the aforementioned method,
is
In certain instances, the present invention relates to the aforementioned method,
i is
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, v' is 2, PJ-
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B
A
2 is , R
3 is alkyl, v
2 is
is
In certain instances, the present invention relates to the aforementioned method,
is
P , and said polymerization agent is ultraviolet light or visible light.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
A
2 is
, R
3 is alkyl, v
2 i is
In certain instances, the present invention relates to the aforementioned method,
visible light.
In certain instances, the present invention relates to the aforementioned method, wherein p
1 is 2.
In certain instances, the present invention relates to the aforementioned method, wherein m is 1. In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, and R3 is (Ci-Cs)alkyl.
In certain instances, the present invention relates to the aforementioned method, wherein p1 is 2, p2 is 0, and R3 is (Ci-Cs)alkyl, and w2 is an integer in the range of about 60 to about 90. In certain instances, the present invention relates to the aforementioned method,
m
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is H, B is
, m
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R i i .
•s„ H, B is
, A
2 , m
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula II. In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1"111 is -CH2C(O)H, and R2 111 Js H. In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1"111 is -CH2C(O)H, R2'1'1
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1"111 is -CH2C(O)H, R2 iπ
is H, B
1'1" is
and w is an integer in the range of about 60- 90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1"111 is -(C(R2'
or
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1'1" is -(C(R2'
)
2)
XC(O)R ' or -C(O)(C(R
2-
m)
2)
yC(O)R
3-
iπ, R
2"1" is H, R
3'1" is
or
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula III, R1"1" is -(C(R2'
UI)
2)
xC(O)R
3'πι or -C(O)(C(R
2'III)
2)
yC(O)R
3'111, R
2'111 is H, R
3'111 is
O or
B
wπ is
and w is an integer in the range of about 60-90.
In certain instances, the present invention relates to the aforementioned method, wherein said compound of formula Ia is
n is an integer in the range of about 70 to about 80, and said polymerization agent is UV light.
In certain embodiments, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula Ib.
In certain embodiments, the present invention relates to the aforementioned method, wherein v is 2.
In certain embodiments, the present invention relates to the aforementioned method, wherein X5 is -N(H)-. In certain embodiments, the present invention relates to the aforementioned method, wherein R18 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R19 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R20 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 20-500.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 40-250. In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 60-90.
In certain embodiments, the present invention relates to the aforementioned method, said compound of formula Ib is
In certain embodiments, the present invention relates to the aforementioned method, said polymerization agent is a compound of formula V.
In certain embodiments, the present invention relates to the aforementioned method, wherein v is 2. In certain embodiments, the present invention relates to the aforementioned method, wherein X6 is -N(H)-.
In certain embodiments, the present invention relates to the aforementioned method, wherein R24 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R25 is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein R is H.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 20-500. In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 40-250.
In certain embodiments, the present invention relates to the aforementioned method, wherein w is an integer in the range of about 60-90.
In certain embodiments, the present invention relates to the aforementioned method, wherein R23 represents independently for each occurrence
In certain embodiments, the present invention relates to the aforementioned method, wherein R23 represents independently for each occurrence
In certain embodiments, the present invention relates to the aforementioned method, said compound of formula V is
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is an oxidizing agent.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is O2.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is ultraviolet light or visible light. In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is ultraviolet light.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is light with a λ of 400-600 nm.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is light with a λ of 450-550 nm.
In certain embodiments, the present invention relates to the aforementioned method, wherein said polymerization agent is light with a λ of 488-514 nm.
In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a primate, bovine, equine, feline, or canine. In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a human.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear, strain, void, fibrillation, or a decrease in the amount of cartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear.
In certain embodiments, the present invention relates to the aforementioned method, wherein said tear is less than about 5 mm long.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in articular cartilage. In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in fibrocartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in the meniscus.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 10 cm2 in size.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 5 cm2 in size.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 1 cm2 in size. In certain embodiments, the present invention relates to the aforementioned method, wherein said compound of formula Ia is dissolved in at least one solvent, and said compound of formula Ia has a concentration in the range of about 2% w/w to about 40% w/w.
In certain embodiments, the present invention relates to the aforementioned method, wherein said compound of formula Ia is dissolved in at least one solvent, and said compound of formula Ia has a concentration in the range of about 5% w/w to about 20% w/w.
In certain embodiments, the present invention relates to the aforementioned method, wherein said compound of formula Ia is dissolved in at least one solvent, and said compound of formula Ia has a concentration in the range of about 6% w/w to about 10% w/w. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing a photoinitiator with said compound of formula Ia prior to exposing said compound of formula Ia to said polymerization agent.
In certain embodiments, the present invention relates to the aforementioned method, wherein said photoinitiator is eosin-Y. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing a natural polymer with said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, wherein said natural polymer is HA, collagen, or a GAG fragment. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing at least one cell with said dendrimeric compound or said repair agent.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cell is a cartilage cell or a stem cell. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent. In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent, wherein said polymerization agent is selected from the group consisting of a compound of formula II, a compound of formula III, a compound of formula IV, and a compound of formula V.
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is performed by treatment with ethylene oxide, hydrogen peroxide, heat, gamma irradiation, electron beam irradiation, microwave irradiation, or visible light irradiation. In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about 10"
3
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about 10" 6.
Another aspect of the present invention relates to a method of repairing cartilaginous tissue, comprising the steps of:
applying an effective amount of a dendrimeric compound of formulae VI , VII, VIII, or IX to a cartilage defect of a patient and exposing said dendrimeric compound to a polymerization agent sufficient to polymerize said dendrimeric compound, wherein said polymerization agent is an oxidizing agent or a compound of formula X, wherein formula VI is represented by:
VI or a pharmaceutically acceptable salt, solvate, or hydrate thereof, wherein
R1 represents independently for each occurrence H, OH, -(C(R3)2)mN(R2)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
R2 represents independently for each occurrence H or alkyl; R3 represents independently for each occurrence H, halogen, or alkyl; R4 represents independently for each occurrence alkyl, aryl, or aralkyl;
R5 represents independently for each occurrence OH, -(C(R3)2)mN(R2)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; and p is 1, 2, 3, 4, or 5; formula VII is represented by:
R
1 represents independently for each occurrence H, -(C(R
3)
2)
mN(H)R
4, - (C(R
3)
2)
mN(R
4)OH, -(C(R
3)
2)
mSH, -C(O)(C(R
3)
2)
mSH, -CO
2(C(R
3)
2)
mSH, -
C(O)N(R
2)(C(R
3)
2)
mSH,
Or
R2 represents independently for each occurrence H, alkyl, or -(C(R3)2)XOR' ; R3 represents independently for each occurrence H, halogen, or alkyl; R4 represents independently for each occurrence H, alkyl, aryl, or aralkyl;
R5 represents independently for each occurrence OH, -(C(R3)2)mN(R2)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 1, 2, 3, 4, or 5; x represents independently for each occurrence 1, 2, 3, or 4; and y is O, 1, 2, 3, or 4; formula VIII is represented by:
VIII wherein
R
1 represents independently for each occurrence H, -(C(R )
2)
mSH, -
R3 N-R4
C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH, R4
R2 represents independently for each occurrence H, alkyl, -(C(R3)2)mYR1, OH, - (C(R3)2)mN(H)R4, -(C(R3)2)mN(R4)OH, -(C(R3)2)mSH, -C(O)(C(R3)2)mSH, -
CO
2(C(R
3)
2)
mSH, -C(O)N(R
2)(C(R
3)
2)
mSH,
or R3 represents independently for each occurrence H, halogen, or alkyl; R represents independently for each occurrence H, alkyl, aryl, or aralkyl; R5 represents independently for each occurrence OH, -(C(R3)2)mN(R2)OH, -
(C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
Y represents independently for each occurrence O or NR
4; n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 1, 2, 3, 4, or 5; and x represents independently for each occurrence 1 , 2, 3, or 4; formula IX is represented by:
IX wherein
R1 represents independently for each occurrence H, -(C(R3)2)mN(H)R4, -
(C(R3)2)mN(R4)OH, -(C(R3)22))mmSSHH,, - -CC((OO))((CC(R3)2)mSH, -CO2(C(R3)2)mSH, -
C(O)N(R2)(C(R3)2)mSH,
R represents independently for each occurrence alkyl, aryl, or aralkyl; R3 represents independently for each occurrence H, halogen, or alkyl; R4 represents independently for each occurrence H, alkyl, aryl, or aralkyl;
R5 represents independently for each occurrence OH, -(C(R3)2)mN(R4)OH, - (C(R3)2)mSH, -C(O)(C(R3)2)mSH, -CO2(C(R3)2)mSH, -C(O)N(R2)(C(R3)2)mSH,
n and m each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, or 8; p represents independently for each occurrence 1, 2, 3, 4, or 5; and x is 1 or 2; and formula X is represented by:
R1-X B_ R1-X
X
wherein
R1 X represents independently for each occurrence -(C(R2'X)2)XC(O)H, -C(O)(C(R2" x)2)yC(0)H, -(C(R2-χ)2)xC(O)R3"x, -C(O)(C(R2-χ)2)yC(O)R3"x, -(C(R2-χ)2)xR4"x, -
R ,2- 'X represents independently for each occurrence H, alkyl, or halogen;
R3" represents independently for each occurrence alkyl, fluoroalkyl, chloroalkyl, -
-R5-X
R4'x represents independently for each occurrence -N=C=O, -N=C=S, R5'x R5'x ,
^,R5-χ
N
-R5-X or ' R5"x R5'X ;
R5"x represents independently for each occurrence H, alkyl, or aralkyl;
B is alkyl diradical, heteroalkyl diradical, or
v
\r . v
2"x represents independently for each occurrence 2, 3, or 4; w
2"x is an integer in the range of about 5 to 1000, inclusive; and x and y each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, 8, or 9.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is an oxidizing agent.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is O2.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X.
In certain instances, the present invention relates to the aforementioned method, w ,2- is an integer in the range of about 50 to about 250.
In certain instances, the present invention relates to the aforementioned method, w " x is an integer in the range of about 60 to about 90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R1 X is -CH2C(O)H, and R2"x is H.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R'"x is -CH2C(O)H, R2'x is
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R*"x is -CH2C(O)H, R2"x is
H, B is
y
2-x j
s 2
} an(j
w 2-x j
s an integer j
n me range of about 15-90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R'"X is -(C(R2'X)2)XC(O)R3'
or -C(O)(C
or
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R
l x is -(C(R
2"X)
2)
XC(O)R
3 In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, R'"x is -(C(R2"X)2)XC(O)R3~
x or -C(O)(C(R
2 x)
2)
yC(O)R
3-
χ, R
2 X is H, R
3-
χ is
B is
is 2, and w
2"x is an integer in the range of about 15-90.
In certain instances, the present invention relates to the aforementioned method, wherein said polymerization agent is a compound of formula X, wherein B is
d
2"x, e
2 X, and g
2'x represent independently an integer greater than zero, provided that the sum of d
2"x, e
2"x, and g
2'x is an integer in the range of about 5 to 1000, inclusive.
In certain instances, the present invention relates to the aforementioned method, wherein, RNX is -(C(R2"X)2)XC(O)R3"X or -C(O)(C(R2'x)2)yC(O)R3-χ, R2-χ is H, and R3"x is
In certain instances, the present invention relates to the aforementioned method,
wherein, formula X is
and s is an integer in the range of about 1-20, inclusive.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI.
In certain instances, the present invention relates to the aforementioned method, wherein n is 3, 4, or 5.
In certain instances, the present invention relates to the aforementioned method, wherein n is 4. In certain instances, the present invention relates to the aforementioned method, wherein R2 is H.
In certain instances, the present invention relates to the aforementioned method, wherein R3 is H.
In certain instances, the present invention relates to the aforementioned method, wherein R4 is alkyl.
In certain instances, the present invention relates to the aforementioned method, wherein R4 is methyl or ethyl.
In certain instances, the present invention relates to the aforementioned method, wherein n is 4, R2 and R3 is H, and R4 is alkyl. In certain instances, the present invention relates to the aforementioned method,
R3 N-R2 wherein R1 is R2
In certain instances, the present invention relates to the aforementioned method,
R3 N-R2 wherein R1 is R2 , and p is 1.
In certain instances, the present invention relates to the aforementioned method,
In certain instances, the present invention relates to the aforementioned method,
wherein R
1 is
and p is 1.
In certain instances, the present invention relates to the aforementioned method,
wherein n is 4, R
2 and R
3 are H, R
4 is methyl, R
1 is
R
2 , and p is 1.
In certain instances, the present invention relates to the aforementioned method,
wherein n is 4, R
2 and R
3 are H, R
4 is methyl, R
1 is
and p is 1.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and a Bronsted acid. In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and HA, wherein A is halogen or -O2CR6, and R6 is alkyl, fluoroalkyl, aryl, or aralkyl.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said
pharmaceutically acceptable salt is a complex formed by said compound of formula VI and an acid selected from group consisting of HCl and HBr.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and HO2CR6, wherein R6 is fluoroalkyl.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VI, and said pharmaceutically acceptable salt is a complex formed by said compound of formula VI and CF3CO2H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VII.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VII, x and y are 1 , R is - CH2OR1, and R3 is H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VII, x is 1 , y is O, and R2 and R3 are H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VIII.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VIII, x is 2, Y is O, R2 is - CH2CH2OR1, and R3 is H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula VIII, x is 2, Y is NR4, and R2 and R3 are H.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula IX.
In certain instances, the present invention relates to the aforementioned method, wherein said dendrimeric compound is a compound of formula IX, R2 is methyl, and x is 2.
In certain instances, the present invention relates to the aforementioned method, further comprising the step of exposing said dendrimeric compound to a compound of formula XI, wherein formula XI is represented by:
R1-XI_BXI_R1-XI
XI
wherein
2-
R*'XI represents independently for each occurrence -(C(R2'XI)2)XC(O)H, -C(O)(C(R' xl)2)yC(O)H, -(C(R2-XI)2)XC(O)R3-XI, -C(O)(C(R2-χi)2)yC(O)R3 XI, -(C(R2"XI)2)xR4-χi, -
R »2- "XI represents independently for each occurrence H, alkyl, or halogen;
R3"XI represents independently for each occurrence alkyl, fluoroalkyl, chloroalkyl, -
R ,4- "XI represents independently for each occurrence -N=C=O, -N=C=S,
R5-Xl
N'
/ \ -R5-χι .R5-Xl R5-XI R5-XI or R'5-XI R5-XI .
R ' represents independently for each occurrence H, alkyl, or aralkyl;
B ,xi is alkyl diradical, heteroalkyl diradical, or
v
2"XI represents independently for each occurrence 2, 3, or 4; w
" is an integer in the range of about 5 to 1000, inclusive; and x and y each represent independently for each occurrence 1, 2, 3, 4, 5, 6, 7, 8, or 9.
In certain embodiments, the present invention relates to the aforementioned method,
wherein, B XI is
and v
2-
xl is 2.
In certain embodiments, the present invention relates to the aforementioned method,
wherein, B XI is
w
2*
1 , v
2-: Xl is 2, R
1 XI is -(C(R 2-XI WXO)R 3-XI
In certain embodiments, the present invention relates to the aforementioned method,
wherein, B X
λI
l is
w
2"*
1 s γ
2-x
i is 2, R'-
χi is -(C(R
2"XI)
2)
XC(O)R 3-XI
or -C(O)(C
is or
said polymerization agent is a compound of formula X, B is
and d
2'x , e
2 x, and g
2"x represent independently an integer greater than zero, provided that the sum of d
2"x, e
20C, and g
2"x is an integer in the range of about 5 to 1000, inclusive.
In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a primate, bovine, equine, feline, or canine.
In certain embodiments, the present invention relates to the aforementioned method, wherein said patient is a human.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear, strain, void, fibrillation, or a decrease in the amount of cartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is a tear.
In certain embodiments, the present invention relates to the aforementioned method, wherein said tear is less than about 5 mm long. In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in articular cartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in fibrocartilage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is an abnormality in the meniscus.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 10 cm in size.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 5 cm2 in size. In certain embodiments, the present invention relates to the aforementioned method, wherein said cartilage defect is less than about 1 cm2 in size.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing a natural polymer with said compound of formula
V.
In certain embodiments, the present invention relates to the aforementioned method, wherein said natural polymer is HA, collagen, or a GAG fragment.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of admixing at least one cell with said dendrimeric compound or said repair agent.
In certain embodiments, the present invention relates to the aforementioned method, wherein said cell is a cartilage cell or a stem cell.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent.
In certain embodiments, the present invention relates to the aforementioned method, further comprising the step of sterilizing said dendrimeric compound and said polymerization agent, wherein said polymerization agent is a compound of formula X.
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is performed by treatment with ethylene oxide, hydrogen peroxide, heat, gamma irradiation, electron beam irradiation, microwave irradiation, or visible light irradiation.
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about 10"3.
In certain embodiments, the present invention relates to the aforementioned method, wherein said sterilizing is effective to achieve a sterility assurance level of at least about IQ-6.
Another aspect of the invention relates to the polymeric cartilage repair composition formed using any one of the above methods.
Another aspect of the invention relates to a method for preparing a biocompatible gel, comprising the step of: admixing a first biocompatible crosslinking polymer comprising at least two different nucleophilic groups with a second biocompatible crosslinking polymer comprising at least one amine-reactive group and at least one sulfhydryl-reactive group to form a gel, wherein said amine- and sulfhydryl-reactive groups are capable of a covalent reaction with said nucleophilic groups of said first crosslinking polymer.
In certain embodiments, the present invention relates to the aforementioned method, wherein said gel is formed in less than about one hour following admixing of said first biocompatible polymer with said second biocompatible polymer.
In certain embodiments, the present invention relates to the aforementioned method, wherein said nucleophilic groups on said first biocompatible polymer are sulfhydryl and amine.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed ex vivo.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vitro.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vivo.
In certain embodiments, the present invention relates to the aforementioned method further comprising the step of administering an effective amount of said gel to a patient.
Another aspect of the invention relates to a method for preparing a biocompatible gel, comprising the step of: admixing a first biocompatible crosslinking polymer comprising at least two different
nucleophilic groups with a second biocompatible crosslinking polymer comprising at least one sulfhydryl-reactive group to form a gel, wherein said sulfhydryl-reactive group is capable of a covalent reaction with said nucleophilic groups of said first crosslinking polymer. In certain embodiments, the present invention relates to the aforementioned method, wherein said gel is formed in less than about one hour following admixing of said first biocompatible polymer with said second biocompatible polymer.
In certain embodiments, the present invention relates to the aforementioned method, wherein said nucleophilic groups on said first biocompatible polymer are sulfhydryl and amine.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed ex vivo.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vitro. In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vivo.
In certain embodiments, the present invention relates to the aforementioned method further comprising the step of administering an effective amount of said gel to a patient.
Another aspect of the invention relates to a method for preparing a biocompatible gel, comprising the step of: admixing a first biocompatible crosslinking polymer comprising at least one amine group and at least one sulfhydryl group with a second biocompatible crosslinking polymer comprising at least one aldehyde to form a gel, wherein said amine group and said sulfhydryl group are capable of covalent reaction with the aldehyde group to form a thiazolidine linkage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said gel is formed in less than about one hour following admixing of said first
biocompatible polymer with said second biocompatible polymer.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed ex vivo.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vitro.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vivo.
In certain embodiments, the present invention relates to the aforementioned method further comprising the step of administering an effective amount of said gel to a patient.
Another aspect of the invention relates to a method for preparing a biocompatible gel, comprising the step of: admixing a first biocompatible crosslinking polymer comprising a histidine amino acid group with a second biocompatible crosslinking polymer comprising an electrophilic group to form a gel, wherein said histidine group of said first polymer and said electrophilic group of said second polymer are capable of reaction to an amide linkage.
In certain embodiments, the present invention relates to the aforementioned method, wherein said electrophilic group is a thiocarboxylic acid or acyl disulfide.
In certain embodiments, the present invention relates to the aforementioned method, wherein said gel is formed in less than about one day following admixing of said first biocompatible polymer with said second biocompatible polymer.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed ex vivo.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vitro.
In certain embodiments, the present invention relates to the aforementioned method, wherein said method is performed in vivo.
In certain embodiments, the present invention relates to the aforementioned method further comprising the step of administering an effective amount of said gel to a patient.
Kits of the Invention One aspect of the present invention relates to a kit for the repairing cartilaginous tissue comprising: a dendrimeric compound of formula Ia or formula Ib, wherein formula Ia and formula Ib are as defined above; and instructions for using said kit. In certain embodiments, the present invention relates to the aforementioned kit, further comprising a desiccant.
In certain embodiments, the present invention relates to the aforementioned kit, further comprising an inert atmosphere to prevent reaction of said dendrimeric compound with atmospheric molecules. In certain embodiments, the present invention relates to the aforementioned kit, further comprising a polymerization agent.
In certain embodiments, the present invention relates to the aforementioned kit, wherein said polymerization agent is a compound of formula II, a compound of formula III, a compound of formula IV, or a compound of formula V; wherein formulae II, III, IV, and V are as defined above.
In certain embodiments, the present invention relates to the aforementioned kit, wherein said kit has a sterility assurance level of at least about 10"3.
In certain embodiments, the present invention relates to the aforementioned kit, wherein said kit has a sterility assurance level of at least about 10"6.
Another aspect of the present invention relates to a kit for the repairing cartilaginous tissue comprising:
a dendrimeric compound of formulae VI, VTI, VIII, or IX, wherein formulae VI, VII, VIII, and IX are as defined above; and instructions for using said kit.
In certain embodiments, the present invention relates to the aforementioned kit, further comprising a desiccant.
In certain embodiments, the present invention relates to the aforementioned kit, further comprising an inert atmosphere to prevent reaction of said dendrimeric compound with atmospheric molecules.
In certain embodiments, the present invention relates to the aforementioned kit, further comprising a polymerization agent.
In certain embodiments, the present invention relates to the aforementioned kit, wherein said polymerization agent is a compound of formula X; and formula X is as defined above.
In certain embodiments, the present invention relates to the aforementioned kit, wherein said kit has a sterility assurance level of at least about 10'3.
In certain embodiments, the present invention relates to the aforementioned kit, wherein said kit has a sterility assurance level of at least about 10"6.
Definitions For convenience, certain terms employed in the specification, examples, and appended claims are collected here.
The term "generation" refers to the number of branched repeat units which emanate from the central core. For example a third generation (or G3) PGLSA dendrimer has three branching layers not including the core. The term "polymerize" as used herein refers to the process of converting a monomer to a chain of momomers, wherein the chain of momomers comprises at least about 5 monomers. In certain instances, the chain of monomers comprises at least about 10 or 15 momomers. In certain instances, the chain of monomers comprises at least about 25 or 40
momomers. In certain instances, the chain of monomers comprises at least about 50 or 75 momomers. In certain instances, the chain of monomers comprises at least about 100 or 150 momomers. In instances wherein the monomeric unit has more than one functional group capable of forming a bond in the polymerization reaction, the term "polymerize" indicates that at least one of the functional groups capable of forming a bond in the polymerization reaction forms a bond with another compound, generally speaking, the other compound is another monomer. In certain instances, at least about 10% of the functional groups capable of forming a bond in a polymerization reaction form a bond to another monomer. In certain instances, at least about 25% of the functional groups capable of forming a bond in a polymerization reaction form a bond to another monomer. In certain instances, at least about 50% of the functional groups capable of forming a bond in a polymerization reaction form a bond to another monomer. In certain instances, at least about 75% of the functional groups capable of forming a bond in a polymerization reaction form a bond to another monomer. In certain instances, about 20% to about 50% of the functional groups capable of forming a bond in a polymerization reaction form a bond to another monomer. Furthermore, the term "polymerize" only requires that at least some of the monomer units in a given solution react to form a chain of monomers. In certain instances, about 10% to about 30% of the monomers react to form a chain of monomers. In certain instances, about 30% to about 50% of the monomers react to form a chain of monomers. In certain instances, about 50% to about 75% of the monomers react to form a chain of monomers. In certain instances, about 75% to about 85% of the monomers react to form a chain of monomers. In certain instances, about 85% to about 95% of the monomers react to form a chain of monomers. In certain instances, greater than about 95% of the monomers react to form a chain of monomers. The term "PEG(NHSV refers to a polyethylene glycol having the following functional group attached at both ends of the polymer chain:
PEG(NHS)
2 can be prepared using either of the following methods. In method 1, a polyethylene glycol is subjected to oxidative conditions in order to oxidize the two termini to the corresponding carboxylic acids [HO
2CCH
2O-PEG-OCH
2CO
2H], followed by transformation to the bis(NHS ester). In method 2, PEG(NHS)
2 is prepared by alkylation of the two termini of a polyethylene glycol with acrylonitrile to give NCCH
2CH
2O-PEG-
OCH2CH2CN, followed by hydrolysis to the bis(acid) [HO2CCH2CH2O-PEG-
OCH2CH2CO2H], and then transformation to the bis(NHS ester). Likewise, poly(propylene glycol) may be subjected to either of the two aforementioned methods to give the corresponding PPG(NHS)2 compounds. In a number of embodiments, the PPG(NHS)2 compounds may be used substantially interchangeably with the PEG(NHS)2 compounds.
The term "heteroatom" as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are boron, nitrogen, oxygen, phosphorus, sulfur and selenium.
The term "alkyl" refers to the radical of saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups. In preferred embodiments, a straight chain or branched chain alkyl has 30 or fewer carbon atoms in its backbone (e.g., Cj-Cβo for straight chain, C3-C30 for branched chain), and more preferably 20 or fewer. Likewise, preferred cycloalkyls have from 3-10 carbon atoms in their ring structure, and more preferably have 5, 6 or 7 carbons in the ring structure.
Unless the number of carbons is otherwise specified, "lower alkyl" as used herein means an alkyl group, as defined above, but having from one to ten carbons, more preferably from one to six carbon atoms in its backbone structure. Likewise, "lower alkenyl" and "lower alkynyl" have similar chain lengths. Preferred alkyl groups are lower alkyls. In preferred embodiments, a substituent designated herein as alkyl is a lower alkyl.
The term "aralkyl", as used herein, refers to an alkyl group substituted with an aryl group (e.g., an aromatic or heteroaromatic group).
The terms "alkenyl" and "alkynyl" refer to unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
The term "aryl" as used herein includes 5-, 6- and 7-membered single-ring aromatic groups that may include from zero to four heteroatoms, for example, benzene, anthracene, naphthalene, pyrene, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, triazole, pyrazole, pyridine, pyrazine, pyridazine and pyrimidine, and the like. Those aryl groups having heteroatoms in the ring structure may also be referred to as "aryl heterocycles" or
"heteroaromatics." The aromatic ring can be substituted at one or more ring positions with such substituents as described above, for example, halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, -CF3, -CN, or the like. The term "aryl" also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls. The terms ortho, meta and para apply to 1,2-, 1,3- and 1 ,4-disubstituted benzenes, respectively. For example, the names 1,2-dimethylbenzene and ort/jo-dimethylbenzene are synonymous.
The terms "heterocyclyl" or "heterocyclic group" refer to 3- to 10-membered ring structures, more preferably 3- to 7-membered rings, whose ring structures include one to four heteroatoms. Heterocycles can also be polycycles. Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxathiin, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, oxolane, thiolane, oxazole, piperidine, piperazine, morpholine, lactones, lactams such as azetidinones and pyrrolidinones, sultams, sultones, and the like. The heterocyclic ring can be substituted at one or more positions with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether,
alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
The terms "polycyclyl" or "polycyclic group" refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings". Rings that are joined through non-adjacent atoms are termed "bridged" rings. Each of the rings of the polycycle can be substituted with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -
CF3, -CN, or the like.
As used herein, the term "nitro" means -NO2; the term "halogen" designates -F, -Cl, -Br or -I; the term "sulfhydryl" means -SH; the term "hydroxyl" means -OH; and the term "sulfonyl" means -SO2-. The terms "amine" and "amino" are art-recognized and refer to both unsubstituted and substituted amines, e.g., a moiety that can be represented by the general formula:
-\R or ~ΓR-
9 R wherein R9, R^Q and R' JQ each independently represent a group permitted by the rules of valence. The term "acylamino" is art-recognized and refers to a moiety that can be represented by the general formula:
O —N—LR^
R9 wherein R9 is as defined above, and R'\ \ represents a hydrogen, an alkyl, an alkenyl or -(CH2)m-R.8> where m and Rg are as defined above.
The term "amido" is art recognized as an amino-substituted carbonyl and includes a moiety that can be represented by the general formula:
wherein R9, Rj 0 are as defined above. Preferred embodiments of the amide will not include imides which may be unstable.
The term "alkylthio" refers to an alkyl group, as defined above, having a sulfur radical attached thereto. In preferred embodiments, the "alkylthio" moiety is represented by one of -S-alkyl, -S-alkenyl, -S-alkynyl, and -S-(CH2)m-R8, wherein m and Rg are defined above. Representative alkylthio groups include methylthio, ethyl thio, and the like. The term "carbonyl" is art recognized and includes such moieties as can be represented by the general formula:
wherein X is a bond or represents an oxygen or a sulfur, and R\ \ represents a hydrogen, an alkyl, an alkenyl, -(CH2)
m-Rg or a pharmaceutically acceptable salt, R'\ \ represents a hydrogen, an alkyl, an alkenyl or -(CH2)
m-Rg, where m and Rg are as defined above. Where X is an oxygen and Rj \ or R'\ \ is not hydrogen, the formula represents an "ester". Where X is an oxygen, and Rj \ is as defined above, the moiety is referred to herein as a carboxyl group, and particularly when Rn is a hydrogen, the formula represents a "carboxylic acid". Where X is an oxygen, and R
1 \ \ is hydrogen, the formula represents a "formate". In general, where the oxygen atom of the above formula is replaced by sulfur, the formula represents a "thiolcarbonyl" group. Where X is a sulfur and R\ \ or R'\ \ is not hydrogen, the formula represents a "thiolester." Where X is a sulfur and Rj \ is hydrogen, the formula represents a "thiolcarboxylic acid." Where X is a sulfur and Ri 1' is hydrogen, the formula represents a "thiolformate." On the other hand, where X is a bond, and R
j \ is not hydrogen, the above formula represents a "ketone" group. Where X is a bond, and R\ \ is hydrogen, the above formula represents an "aldehyde" group.
The terms "alkoxyl" or "alkoxy" as used herein refers to an alkyl group, as defined above, having an oxygen radical attached thereto. Representative alkoxyl groups include methoxy, ethoxy, propyloxy, tert-butoxy and the like. An "ether" is two hydrocarbons covalently linked by an oxygen. Accordingly, the substituent of an alkyl that renders that alkyl an ether is or resembles an alkoxyl, such as can be represented by one of -O-alkyl, -O- alkenyl, -0-alkynyl, -O-(CH2)
m-R8
> where m and Rg are described above.
The term "sulfonate" is art recognized and includes a moiety that can be represented by the general formula:
O Il -S-OR41
O in which R41 is an electron pair, hydrogen, alkyl, cycloalkyl, or aryl.
What about PLA, PGA1PLGA, etc.
The terms triflyl, tosyl, mesyl, and nonaflyl are art-recognized and refer to trifluoromethanesulfonyl, /7-toluenesulfonyl, methanesulfonyl, and nonafluorobutanesulfonyl groups, respectively. The terms triflate, tosylate, mesylate, and nonaflate are art-recognized and refer to trifluoromethanesulfonate ester, p-toluenesulfonate ester, methanesulfonate ester, and nonafluorobutanesulfonate ester functional groups and molecules that contain said groups, respectively.
The abbreviations Me, Et, Ph, Tf, Nf, Ts, Ms represent methyl, ethyl, phenyl, trifluoromethanesulfonyl, nonafluorobutanesulfonyl, /7-toluenesulfonyl and methanesulfonyl, respectively. A more comprehensive list of the abbreviations utilized by organic chemists of ordinary skill in the art appears in the first issue of each volume of the Journal of Organic Chemistry; this list is typically presentd in a table entitled Standard List of Abbreviations. The abbreviations contained in said list, and all abbreviations utilized by organic chemists of ordinary skill in the art are hereby incorporated by reference. The term "sulfate" is art recognized and includes a moiety that can be represented by the general formula:
O Il 0-S-OR41
Il O
in which R41 is as defined above.
The term "sulfonylamino" is art recognized and includes a moiety that can be represented by the general formula:
O Il N-S-R
I O
R The term "sulfamoyl" is art-recognized and includes a moiety that can be represented by the general formula:
The term "sulfonyl", as used herein, refers to a moiety that can be represented by the general formula:
O Il S R44 0 in which R44 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aryl, or heteroaryl.
The term "sulfoxido" as used herein, refers to a moiety that can be represented by the general formula:
O -S-R« in which R44 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aralkyl, or aryl.
A "selenoalkyl" refers to an alkyl group having a substituted seleno group attached thereto. Exemplary "selenoethers" which may be substituted on the alkyl are selected from one of -Se-alkyl, -Se-alkenyl, -Se-alkynyl, and -Se-(CH2)m-R7, m and R7 being defined above.
Analogous substitutions can be made to alkenyl and alkynyl groups to produce, for example, aminoalkenyls, aminoalkynyls, amidoalkenyls, amidoalkynyls, iminoalkenyls, iminoalkynyls, thioalkenyls, thioalkynyls, carbonyl-substituted alkenyls or alkynyls.
As used herein, the definition of each expression, e.g. alkyl, m, n, etc., when it occurs more than once in any structure, is intended to be independent of its definition elsewhere in the same structure.
It will be understood that "substitution" or "substituted with" includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc.
As used herein, the term "substituted" is contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds. Illustrative substituents include, for example, those described herein above. The permissible substituents can be one or more and the same or different for appropriate organic compounds. For purposes of this invention, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. This invention is not intended to be limited in any manner by the permissible substituents of organic compounds.
The phrase "protecting group" as used herein means temporary substituents which protect a potentially reactive functional group from undesired chemical transformations. Examples of such protecting groups include esters of carboxylic acids, silyl ethers of alcohols, and acetals and ketals of aldehydes and ketones, respectively. The field of protecting group chemistry has been reviewed (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991).
Certain compounds of the present invention may exist in particular geometric or stereoisomeric forms. The present invention contemplates all such compounds, including cis- and frarcs-isomers, R- and S-enantiomers, diastereomers, (D)-isomers, (L)-isomers, the
racemic mixtures thereof, and other mixtures thereof, as falling within the scope of the invention. Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention. If, for instance, a particular enantiomer of a compound of the present invention is desired, it may be prepared by asymmetric synthesis, or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers. Alternatively, where the molecule contains a basic functional group, such as amino, or an acidic functional group, such as carboxyl, diastereomeric salts are formed with an appropriate optically-active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means well known in the art, and subsequent recovery of the pure enantiomers.
Contemplated equivalents of the compounds described above include compounds which otherwise correspond thereto, and which have the same general properties thereof
(e.g., functioning as analgesics), wherein one or more simple variations of substituents are made which do not adversely affect the efficacy of the compound in binding to sigma receptors. In general, the compounds of the present invention may be prepared by the methods illustrated in the general reaction schemes as, for example, described below, or by modifications thereof, using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants which are in themselves known, but are not mentioned here.
The term "alkali metal" refer to those elements listed in Group 1 of the periodic table. The following elements are alkali metals: Li, Na, K, Rb, Cs, and Fr. For purposes of this invention, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 67th Ed., 1986-87, inside cover.
Exemplification
The invention now being generally described, it will be more readily understood by reference to the following examples, which are included merely for purposes of illustration of certain aspects and embodiments of the present invention, and are not intended to limit the invention.
Example 1
Synthesis of 2-[(cis-l,3-benzylidene glycerol)-2-propionic acid] - c/s-l,3-OBenzylidene glycerol (10.9 g, 60.4 mmol) was dissolved in 1,4-dioxane (250 mL) followed by the addition of NaH (7.0 g, 0.30 mol). The reaction mixture was stirred at rt for one hour before cooling to 0 0C. 2-Bromopropionic acid (8.64 mL, 96 mmol) was then added over a 15 minute period of time. The reaction mixture was allowed to return to rt and then stirred at 50 0C for 12 hours before it was cooled to 0 0C and quenched with ethanol followed by the addition of water (250 mL). The solution was adjusted to 4.0 pH using IN HCl and extracted with CH2Cl2 (200 mL). This procedure was repeated once again after re-adjusting the pH to 4.0. The combined organic phase was dried with Na2SO4, gravity filtered, and evaporated. The solid was stirred in ethyl ether (50 mL) for 45 minutes and cooled to -25 0C for 3 hours before collecting 11.7 g of the white powder (77.3 % yield). 1H NMR (400 MHz, CDCl3): δ 1.51 (d, 3, CH-CH3, J=7.00 Hz), 3.46 (m, 1, -CH2-CH-CH2-, J=1.71 Hz), 4.04 (m, 2, -CH2-CH-CH21, J=1.71 Hz), 4.22 (q, 1, CH-CH3, J= 7.00 Hz), 4.29 (m, 2, -CH2- CH-CH21, J=I.71 Hz), 5.54 (s, 1, CH), 7.34 (m, 3, arom. CEQ17.46 (m, 2, arom. CH). 13C NMR (400 MHz, CDCl3): δ 176.05 (COOH), 137.82 (CH), 129.34 (CH), 128.52 (CH), 126.26 (CH), 101.79 (CH), 72.83 (CH), 70.70 (CH), 69.28 (CH2), 69.09 (CH2), 18.79 (CH3). FTIR: v (cm 1) 1714 (C=O), 1455 (CH2 bend), 1401 (CH3 bend). GC-MS 253 m/z (MH+) (Theory: 252 m/z (M+)). GC-MS 253 m/z (MH+) (Theory: 252 m/z (M+)) Elemental Analysis C: 61.75 %; H 6.37 % (Theory: C: 61.90 %; H 6.39 %).
Example 2
Synthesis of benzylidene protected [GO]-PGLLA-bzld - 2-[(czs-l,3-benzylidene glycerol)-2 -propionic acid] (4.02 g, 15.9 mmol), cw-l,3-O-benzylideneglycerol (2.62 g, 14.5 mmol), and DPTS (1.21 g, 4.10 mmol) were dissolved in CH2Cl2 (40 mL). The
reaction flask was flushed with nitrogen and then DCC (3.61 g, 17.5 mmol) was added. Stirring at room temperature was continued for 14 hours under a nitrogen atmosphere. Upon reaction completion, the DCC-urea was filtered and washed with a small amount of CH2Cl2 (10 raL) and the filtrate was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 MeOH:CH2Cl2. The product was dissolved in minimal CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Ethyl ether was decanted and the precipitate was exposed to reduced pressure to yield 5.63 g of a white powder (94.0 % yield). 1H NMR (400 MHz, CDCl3): δ 1.56 (d, 6, CH-CH3, J=6.84 Hz), 3.47 (m, 2, -CH2-CH-CH2-, J=1.71 Hz), 3.99 (m, 2, -CH2-CH-CH21, J=I .71 Hz), 4.14 (m, 2, -CH2-CH-CH21, J=I .71 Hz), 4.25 (m, 2, - CH2-CH-CH21), 4.31 (m, 1, -CH2-CH-CH21), 4.37 (q, 1, CH-CH3, J= 6.84 Hz), 4.42 (m, 1, - CH2-CH-CH21), 4.72 (m, 1, -CH2-CH-CH2-, J=1.71 Hz), 5.49 (s, 1, CH), 5.53 (s, 1, CH), 7.34 (m, 6, arom. CH). 7.47 (m, 4, arom. CH). 13C NMR (400 MHz, CDCl3): δ 173.53 (COOR), 138.32 (CH), 137.97 (CH), 129.36 (CH), 129.10 (CH), 128.54 (CH), 128.40 (CH), 126.42 (CH), 126.20 (CH), 101.51 (CH), 101.46 (CH), 72.88 (CH), 70.80 (CH2), 70.23 (CH), 69.08 (CH2), 69.02 (CH2), 68.19 (CH2), 66.83 (CH), 19.34 (CH3). FTIR: v (cm"1) 1743 (C=O), 1452 (CH2 bend), 1389 (CH3 bend). GC-MS 415 m/z (MH+) (Theory: 414 m/z (M+)) Elemental Analysis C: 66.63 %; H 6.33 % (Theory C: 66.65 %; H 6.32 %).
Example 3 Synthesis of [GO]-PGLLA-OH - Pd/C (10%) (10 % w/w) was added to a solution of benzylidene protected [GO]-PGLLA (5.49 g, 13.2 mmol) in EtOAc/MeOH (3:1, 40 mL). The flask was evacuated and filled with 50 psi Of H2 before shaking for 20 minutes. The catalyst was filtered and washed with EtOAc (10 mL). The filtrate was then evaporated to give 2.94 g of a colorless, viscous oil (94.0 % yield). 1H NMR (400 MHz, (CD3)2CO): δ 1.08 (m, 1, CH3), 1.36 (m, 2, CH3), 3.65 (broad m, 9, -CH2-CH-CH2-), 4.20 (broad m, 3, - CH2-CH-CH2-). 13C NMR (400 MHz, (CD3)2SO): δ 174.03 (COOR), 81.53 (CH), 76.66 (CH), 74.30 (CH), 61.82 (CH2), 61.69 (CH2), 60.37 (CH2), 19.62 (CH3). FTIR: v (cml) 3383 (OH), 1737 (C=O). GC MS 239 m/z (MH+) (Theory: 238 m/z (M+)) Elemental Analysis C: 45.52 %; H 7.65 % (Theory C: 45.37 %; H 7.62%).
Example 4
Synthesis of benzylidene protected [Gl]-PGLLA-bzld - 2-[(cw-l,3-benzylidene glycerol)-2-propionic acid] (4.41 g, 17.50 mmol), [GO]-PGLLA (0.791 g, 3.32 mmol), and DPTS (2.46 g, 8.36 mmol), were dissolved in DMF (80 mL). The reaction flask was flushed with nitrogen and then DCC (5.31 g, 25.74 mmol) was added. The contents were stirred at room temperature for 14 hours under nitrogen atmosphere. The DMF was removed under high vacuum and the remaining residue was dissolved in CH2Cl2. The DCC-urea was filtered and washed with a small amount of CH2Cl2 (20 mL) and the filtrate was concentrated. The crude product was purified by silica gel chromatography, eluting with 3 :97 MeOH:CH2Cl2. The product was dissolved in minimal CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Ethyl ether was decanted and the precipitate was exposed to reduced pressure to yield 3.45 g of a white powder (88.3 % yield). 1H NMR (400 MHz, CDCl3): δ 1.33 (m, 3, CH3), 1.47 (m, 12, CH3), 3.41 (m, 4, CH,), 3.76 (m, 2, -CH2-CH-CH2-), 3.97 (m, 4, -CH2-CH-CH2-), 4.10 (m, 4, -CH2-CH-CH2-), 4.28 (m, 20, -CH2-CH-CH2-), 5.30 (m, 1, CH), 5.49 (m, 4, CH), 7.30 (m, 12, arom. CH), 7.46 (m, 8, arom. CH). 13C NMR (400 MHz, CDCl3): δ 173.16 (COOR), 138.24 (CH), 129.14 (CH), 128.40 (CH), 126.36 (CH), 101.47 (CH), 72.68 (CH), 70.54 (CH2), 70.12 (CH), 68.13 (CH2), 19.27 (CH3), 18.99 (CH3). FTIR: v ( cm-1) 1745 (C=O), 1451 (CH2 bend), 1386 (CH3 bend). FAB MS 1175.6 m/z (MH+) (Theory: 1175.2 m/z (M+)) Elemental Analysis C: 62.11 %; H 6.46 % (Theory C: 62.34 %; H 6.35%). SEC Mw: 1280, Mn: 1260, PDI: 1.01.
Example 5
Synthesis of [Gl]-PGLLA-OH - Pd/C (10%) (10 % w/w) was added to a solution of benzylidene protected [Gl]-PGLLA (0.270 g, 0.230 mmol) in THF (15 mL). The flask was evacuated and filled with 50 psi Of H2 before shaking for 15 minutes. The catalyst was filtered and washed with THF (10 mL). The filtrate was then evaporated to give 0.178 g of a colorless, viscous oil (94.0 % yield). 1H NMR (400 MHz, (CD3)2CO): δ 1.41 (m, 5, CH3), 1.49 (m, 10, CH3), 3.53 (m, 2, -CH2-CH-CH2-), 3.63 (m, 11, -CH2-CH-CH2-), 3.74 (m, 4, -CH2-CH-CH2-), 3.93 (m, 3, -CH2-CH-CH2-), 4.23 (m, 5, -CH2-CH-CH2-), 4.39 (m, 10, -CH2-CH-CH2-). 13C NMR (400 MHz, CD3Cl): δ 169.64 (COOR), 74.53 (CH), 72.97
(CH), 72.74 (CH), 69.95 (CH2), 68.97 (CH), 62.73 (CH2), 61.76 (CH2), 19.42 (CH3), 18.13 (CH3), 17.56 (CH3). FTIR: v (cm0) 3409 (OH), 1733 (C=O), 1453 (CH2 bend), 1374 (CH3 bend). FAB MS 823.3 m/z (MH+) (Theory: 822.8 m/z (M+)) Elemental Analysis C: 47.72 %; H 7.41 % (Theory C: 48.17 %; H 7.11 %). SEC Mw: 1100, Mn: 1090, PDI: 1.01.
Example 6
Synthesis of benzylidene protected [G2]-PGLLA-bzld - 2-[(cw-l,3-benzylidene glycerol)-2-propionic acid] (8.029 g, 31.83 mmol), DCC (9.140 g, 44.30 mmol), and DPTS (4.629 g, 15.74 mmol) were dissolved in THF (80 mL). The reaction flask was flushed with nitrogen and stirred for 30 minutes before [Gl]-PGLLA (0.825 g, 1.00 mmol) was added by dissolving in a minimal amount of THF. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. The DCC-urea was filtered and washed with a small amount of THF (20 mL). The THF filtrate was evaporated and the crude product was purified by silica gel chromatography, eluting with 3:97 MeOH:CH2Cl2. The product was dissolved in minimal CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Ethyl ether was decanted and the precipitate was exposed to reduced pressure to yield 2.09 g of a white powder (77 % yield). 1H NMR (400 MHz, CDCl3): δ 1.33 (m, 15, CH3), 1.46 (m, 24, CH3), 3.40 (m, 8, CH,), 3.77 (m, 5, -CH2-CH-CH2-), 3.95 (m, 10, -CH2-CH-CH2-), 4.06 (m, 12, -CH2-CH- CH2-), 4.28 (m, 47, -CH2-CH-CH2-), 5.49 (m, 8, CH), 7.30 (m, 24, arom. CH), 7.47 (m, 16, arom. CH). 13C NMR (400 MHz, CDCl3): δ 173.15 (COOR), 138.28 (CH), 129.12 (CH), 128.40 (CH), 126.36 (CH), 101.44 (CH), 72.69 (CH), 70.54 (CH2), 70.12 (CH), 68.13 (CH2), 19.23 (CH3). FTIR: v ( cm'1) 1746 (C=O), 1452 (CH2 bend), 1386 (CH3 bend). FAB MS 2697.0 m/z (MH+) (Theory: 2696.8 m/z (M+)) Elemental Analysis C: 60.86 %; H 6.37% (Theory C: 61.02 %; H 6.35 %). SEC Mw: 2350, Mn: 2310, PDI: 1.01.
Example 7
Synthesis of [G2]-PGLLA-OH - Pd/C (10%) (10 % w/w) was added to a solution of benzylidene protected [G2]-PGLLA (0.095 g, 0.035 mmol) in THF (10 mL). The flask was evacuated and filled with 50 psi Of H2 before shaking for 15 minutes. The catalyst was filtered and washed with THF (10 mL). The filtrate was evaporated to give 0.061 g of a
colorless viscous oil (88.0 % yield). 1H NMR (400 MHz, (CD3)2CO): δ 1.36 (m, 39, CH3), 3.61 (m, 48, -CH2-CH-CH2-), 3.94 (m, 10, -CH2-CH-CH2-), 4.16 (m, 6, -CH2-CH-CH2-), 4.35 (m, 29, -CH2-CH-CH2-). 13C NMR (400 MHz, (CD3)2CO): δ 174.37 (COOR), 81.98 (CH), 74.16 (CH)1 70.46 (CH), 62.32 (CH2), 62.09 (CH2), 18.76 (CH3). FTIR: v (cm4) 3431 (OH), 1741 (C=O), 1453 (CH2 bend), 1376 (CH3 bend). MALDI-TOF MS 1991.8 m/z (MH+) (Theory: 1991.9m/z (M+)). SEC Mw: 2170, Mn: 2130, PDI: 1.01.
Example 8
Synthesis of [G2]-PGLLA-Ac - [G2J-PGLLA (0.098 g, 0.049 mmol) was dissolved in 5 rnL of pyridine. Acetic anhydride (6.0 mL, 64 mmol) was then added via syringe and the reaction mixture was stirred at 40 0C for 8 hours. Pyridine and acetic anhydride were removed under high vacuum. The product was isolated on a prep TLC eluting with 4:96 MeOH: CH3Cl. 1H NMR (400 MHz, CD3Cl): δ 1.22 (m, 15, CH3), 1.39 (m, 24, CH3), 2.05 (m, 48, CH3), 3.62 - 4.21 (broad multiplets, 83, -CH2-CH-CH2-). 13C NMR (400 MHz, CD3Cl): δ 172.69 (COOR), 170.87 (COOR), 75.15 (CH), 74.60 (CH), 70.46 (CH), 63.68 (CH2), 63.17 (CH2), 29.88 (CH3), 21.02 (CH3), 19.01 (CH3). FAB MS 2665.0 m/z (MH+) (Theory: 2664.5 m/z (M+)) Elemental Analysis C: 50.70 %; H 6.71 % (Theory C: 50.94 %; H 6.43 %).
Example 9 Synthesis of benzylidene protected [G3]-PGLLA-bzld - 2-[(cw-l,3-benzylidene glycerol)-2-propionic acid] (0.376 g, 1.49 mmol), DCC (0.463 g, 2.24 mmol), and DPTS (0.200 g, 0.680 mmol) were dissolved in THF (15 mL). The reaction flask was flushed with nitrogen and stirred for 1.5 hours before [G2]-PGLLA (0.070 g, 0.035 mmol) was added by dissolving in a minimal amount of THF. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. The DCC-urea was filtered and washed with a small amount of THF (20 mL). The THF filtrate was evaporated and the crude product was purified by silica gel chromatography, eluting with 3:97 MeOH:CH2Cl2. The product was dissolved in minimal CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Ethyl ether was decanted and the precipitate was exposed to reduced pressure to yield 0.164 g of a white powder
(89.1 % yield). 1H NMR (400 MHz, CDCl3): δ 1.32 (m, 39, CH3), 1.45 (m, 48, CH3), 3.38 (m, 16, CH,), 3.77 (m, 14, -CH2-CH-CH2-), 3.97 (m, 20, -CH2-CH-CH2-), 4.07 (m, 24, - CH2-CH-CH2-), 4.24 (m, 97, -CH2-CH-CH2-), 4.39 (m, 8, -CH2-CH-CH2-), 5.47 (m, 16, CH), 7.31 (m, 48, arom. CH), 7.44 (m, 32, arom. CH). 13C NMR (400 MHz, CDCl3): δ 173.14 (COOR), 138.28 (CH), 129.12 (CH), 128.40 (CH), 126.36 (CH), 101.41 (CH), 72.68 (CH), 70.56 (CH2), 70.13 (CH), 68.11 (CH2), 19.25 (CH3), 19.02 (CH3). FTIR: v ( cm"1) 1744 (C=O), 1451 (CH2 bend), 1385 (CH3 bend). MALDI MS 5743.3 m/z (MH+) (Theory: 5739.9 m/z (M+)) Elemental Analysis C: 60.32 %; H 6.34% (Theory C: 60.47 %; H 6.36 %). SEC Mw: 4370, Mn: 4310, PDI: 1.01.
Example 10
Synthesis of [G3] -PGLLA-OH - Pd/C (10%) (10 % w/w) was added to a solution of benzylidene protected [G3]-PGLLA (0.095 g, 0.035 mmol) in THF (15 mL). The flask was evacuated and filled with 50 psi Of H2 before shaking for 15 minutes. The catalyst was filtered and washed with THF (10 mL). The filtrate was evaporated to give 0.128 g of a colorless viscous oil (95.4 % yield). 1H NMR (400 MHz, (CD3)2CO): δ 1.37 (m, 87, CH3), 3.56 (m, 83, -CH2-CH-CH2- or -CH-CH3), 3.78 (m, 13, -CH2-CH-CH2- or -CH-CH3), 4.01 (m, 14, -CH2-CH-CH2- or -CH-CH3), 4.18 (m, 13, -CH2-CH-CH2- or -CH-CH3), 4.39 (m, 56, -CH2-CH-CH2- or -CH-CH3). 13C NMR (400 MHz, (CD3)2CO): δ 174.37 (COOR), 82.01 (CH), 74.16 (CH), 62.35 (CH2), 62.15 (CH2), 18.80 (CH3). FTIR: v (cm 1) 3434 (OH), 1738 (C=O), 1452 (CH2 bend), 1376 (CH3 bend). MALDI MS 4332.5 m/z (MH+) (Theory: 4330.2 m/z (M+)) Elemental Analysis C: 49.56 %; H 7.21 % (Theory C: 49.09 %; H 6.94%). SEC Mw: 4110, Mn: 4060, PDI: 1.01.
Example 11
Synthesis of [G0]-PGLSA-bzld - Succinic acid (1.57 g, 13.3 mmol), cis-l,3-O- benzylideneglycerol (5.05 g, 28.0 mmol), and DPTS (4.07 g, 13.8 mmol) were dissolved in CH2Cl2 (120 mL). The reaction flask was flushed with nitrogen and then DCC (8.19 g, 39.7 mmol) was added. Stirring at room temperature was continued for 14 hours under a nitrogen atmosphere. Upon reaction completion, the DCC-urea was filtered and washed with a small amount Of CH2Cl2 (20 mL). The crude product was purified by silica gel
chromatography, eluting with 3:97 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Following vacuum filtration, 5.28 g of a white solid was collected (90 % yield). 1H NMR (CDCl3): δ 2.78 (s, 4, -CH2-CH2-), 4.08 (m, 4, -CH2-CH-CH2-), 4.23 (m, 4, -CH2-CH-CH2-), 4.69 (m, 2, -CH2-CH-CH2-, J=I.54 Hz, 1.71 Hz), 5.50 (s, 2, CH), 7.34 (m, 6, arom. CH), 7.48 (m, 4, arom. CH). 13C NMR (CDCl3): δ 172.32 (COOR), 138.03 (CH), 129.23 (CH), 128.48 (CH), 126.24 (CH), 101.33 (CH), 69.16 (CH2), 66.50 (CH), 29.57 (CH2). FTIR: v (cm"1) 2992 (aliph. C-H stretch), 1727 (C=O). GC-MS 443 m/z (MH+) (Theory: 442 m/z (M+)). HR FAB 442.1635 m/z (M+) (Theory: 442.1628 m/z (M+)). Elemental Analysis C: 65.25 %; H 5.85 % (Theory C: 65.15 %; H 5.92 %).
Example 12
Synthesis of [GO]-PGLSA-OH - Pd/C (10 % w/w) was added to a solution of benzylidene protected [GO]-PGLSA (2.04 g, 4.61 mmol) in THF (30 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 50 psi Of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF (20 mL). The filtrate was evaporated to give 1.18 g of a clear viscous oil (97 % yield). 1H NMR (CD3OD): δ 2.67 (s, 4, -CH2-CH2- ), 3.64 (m, 8, -CH2-CH-CH2-), 4.87 (m, 2, -CH2-CH-CH2-). 13C NMR (CD3OD): D 172.77 (COOR), 75.84 (CH2), 60.41 (CH), 28.96 (CH2). 13C NMR ((CD3)2CO): δ 171.99 (COOR), 76.15 (CH2), 60.89 (CH). FTIR: v (cm'1) 3299 (OH), 1728 (C=O). GC-MS 284 m/z (M+NH4 +) (Theory: 266 m/z (M+)). Elemental Analysis C: 44.94 %; H 6.87 % (Theory C: 45.11 %; H 6.81%).
Example 13
Synthesis of 2-(ci.s-l,3-0-benzylidene glycerol)succinic acid mono ester - cis-l,3-O- Benzylideneglycerol (9.90 g, 54.9 mmol) was dissolved in pyridine (100 mL) followed by the addition of succinic anhydride (8.35 g, 83.4 mmol). The reaction mixture was stirred at room temperature for 18 hours before the pyridine was removed under vacuum at 40 0C. The remaining solid was dissolved in CH2Cl2 (100 mL) and washed three times with cold 0.2 N HCl (100 mL), or until the aqueous phase remained at pH 1. The organic phase was evaporated and the solid was dissolved in deionized water (300 mL). 1 N NaOH was added
until pH 7 was obtained and the product was dissolved in solution. The aqueous phase was extracted with CH2Cl2 (200 raL) and then readjusted to pH 4. The aqueous phase was subsequently extracted twice with CH2Cl2 (200 mL), dried with Na2SO4, filtered, and evaporated. The solid was stirred in ethyl ether (50 mL) and cooled to -25 0C for 3 hours before collecting 14.6 g of a white powder (95 % yield). 1H NMR (CDCl3): δ 2.68 (m, 4, - CH2-CH2-), 4.13 (m, 2, -CH2-CH-CH2-), 4.33 (m, 2, -CH2-CH-CH2-), 4.70 (m, 1, -CH2-CH- CH2-), 5.51 (s, 1, CH), 7.34 (m, 3, arom. CH), 7.47 (m, 2, arom. CH). 13C NMR (CDCl3): δ 178.07 (COOH), 172.38 (COOR), 137.95 (CH), 129.33 (CH), 128.51 (CH), 126.26 (CH), 101.43 (CH), 69.15 (CH2), 66.57 (CH), 29.24 (CH2), 29.05 (CH2). FTIR: v (cm"1) 2931 (aliph. C-H stretch), 1713 (C=O). GC-MS 281 m/z (MH+) (Theory: 280 m/z (M+)). Elemental Analysis C: 60.07 %; H 5.80 % (Theory: C: 59.99 %; H 5.75 %).
Example 14
Synthesis of [Gl]-PGLSA-bzld - 2-(αs-l,3-OBenzylidene glycerol)succinic acid mono ester (6.33 g, 22.6 mmol), [GO]-PGLSA (1.07 g, 4.02 mmol), and DPTS (2.51 g, 8.53 mmol) were dissolved in THF (60 mL). The reaction flask was flushed with nitrogen and then DCC (7.04 g, 34.1 mmol) was added. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount of THF (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 to 5:95 methanol: CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. The ethyl ether was decanted and the precipitate was isolated to yield 5.11 g of a white powder (97 % yield). 1H NMR (CDCl3): δ 2.58 (m, 4, -CH2-CH2-), 2.63 (m, 8, -CH2-CH2-), 2.71 (m, 8, -CH2- CH2-), 4.12 (m, 12, -CH2-CH-CH2-), 4.23 (m, 12, -CH2-CH-CH2-), 4.69 (m, 4, -CH2-CH- CH2-), 5.20 (m, 2, -CH2-CH-CH2-), 5.51 (m, 4, CH), 7.33 (m, 12, arom. CH), 7.46 (m, 8, arom. CH). 13C NMR (CDCl3): δ 172.28 (COOR), 171.91 (COOR), 171.53 (COOR), 138.03 (CH), 129.26 (CH), 128.48 (CH), 126.22 (CH), 101.32 (CH), 69.50 (CH), 69.16 (CH2), 66.54 (CH), 62.49 (CH2), 29.36 (CH2), 29.03 (CH2). FTIR: v (cm 1) 2858 (aliph. C- H stretch), 1731 (C=O). FAB MS 1315.6 m/z (MH+) (Theory: 1315.3 m/z (M+)).
Elemental Analysis C: 60.13 %; H 5.82 % (Theory C: 60.27 %; H 5.67%). SEC Mw: 1460, Mn: 1450, PDI: 1.01.
Example 15
Synthesis of [Gl]-PGLSA-OH - Pd/C (10 % w/w) was added to a solution of benzylidene protected [Gl]-PGLSA (0.270 g, 0.230 mmol) in THF (20 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 50 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF (20 mL). The filtrate was evaporated to give 0.178 g of a colorless, viscous oil (94 % yield). 1H NMR (CD3OD): δ 2.63 (m, 20, - CH2-CH2-), 3.52 (m, 4, -CH2-CH-CH2-), 3.64 (m, 8, -CH2-CH-CH2-), 3.80 (m, 2, -CH2-CH- CH2-), 4.05 (m, 2, -CH2-CH-CH2-), 4.14 (m, 2, -CH2-CH-CH2-), 4.21 (m, 4, -CH2-CH-CH2- ), 4.30 (m, 4, -CH2-CH-CH2-), 4.85 (m, 2, -CH2-CH-CH2-), 5.25 (m, 2, -CH2-CH-CH2-). 13C NMR (CD3OD): δ 172.82 (COOR), 172.58 (COOR), 172.48 (COOR), 172.08 (COOR),
75.82 (CH), 69.90 (CH), 69.68 (CH), 65.66 (CH2), 62.85 (CH2), 62.30 (CH2), 60.43 (CH2),
28.83 (CH2), 28.61 (CH2). FTIR: v (cm'1) 3405 (OH), 2943 (aliph. C-H stretch), 1726 (C=O). FAB MS 963.2 m/z (MH+) (Theory: 962.9 m/z (M+)). Elemental Analysis C:
47.13 %; H 6.11 % (Theory C: 47.40 %; H 6.07 %). SEC Mw: 1510, Mn: 1500, PDI: 1.01.
Example 16
Synthesis of [G2]-PGLSA-bzld - 2-(cιs-l,3-0-Benzylidene glycerol)succinic acid mono ester (4.72 g, 16.84 mmol), [Gl]-PGLSA (1.34 g, 1.39 mmol), and DPTS (1.77 g, 6.02 mmol) were dissolved in THF (100 mL). The reaction flask was flushed with nitrogen and then DCC (4.62 g, 22.4 mmol) was added. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount of THF (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 to 5:95 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. The ethyl ether was decanted and the precipitate was isolated to yield 4.00 g of a white powder (94 % yield). 1H NMR (CDCl3): δ 2.59 (broad m, 26, -CH2-CH2-), 2.69 (broad m, 52, -CH2-CH2-), 4.13 (m, 28, -CH2-CH-CH2-), 4.13 (m, 28, -CH2-CH-CH2-), 4.69 (m, 8, -CH2-CH-CH2-), 5.22
(m, 6, -CH2-CH-CH2-), 5.50 (s, 8, CH), 7.32 (m, 24, arom. CH), 7.47 (m, 16, arom. CH). 13C NMR (CDCl3): δ 172.27 (COOR), 171.88 (COOR), 171.60 (COOR), 138.04 (CH), 129.25 (CH), 128.47 (CH), 126.21 (CH), 101.30 (CH), 69.48 (CH), 69.15 (CH2), 66.54 (CH), 62.57 (CH2), 29.35 (CH2), 29.18 (CH2) 29.03 (CH2), 28.84 (CH2). FTIR: ycm-1) 2969 (aliph. C-H stretch), 1733 (C=O). FAB MS 3060.7 m/z (MH+) (Theory: 3060.9 m/z (M+)). Elemental Analysis C: 59.20 %; H 5.64 % (Theory C: 58.86 %; H 5.60 %). SEC Mw: 3030, Mn: 2990, PDI: 1.01.
Example 17 Synthesis of [G2]-PGLSA-OH - Pd/C (10 % w/w) was added to a solution of benzylidene protected [G2]-PGLSA (2.04 g, 0.667 mmol) in THF (20 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 50 psi Of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF (20 mL). The filtrate was evaporated to give 1.49 g of a colorless, viscous oil (95 % yield). 1H NMR (CD3OD): δ 2.64 (m, 52, - CH2-CH2-), 3.53 (m, 16, -CH2-CH-CH2-), 3.64 (m, 4, -CH2-CH-CH2-), 3.80 (m, 8, -CH2- CH-CH2-), 4.06 (m, 8, -CH2-CH-CH2-), 4.14 (m, 6, -CH2-CH-CH2-), 4.21 (m, 11, -CH2- CH-CH2-), 4.30 (m, 11, -CH2-CH-CH2-), 5.25 (m, 6, -CH2-CH-CH2-). 13C NMR (CD3OD): δ 172.83 (COOR), 172.59 (COOR), 172.49 (COOR), 69.91 (CH), 69.69 (CH), 65.68 (CH2), 62.88 (CH2), 62.37 (CH2), 28.61 (CH2). FTIR: v (cm"1) 3429 (OH), 2952 (aliph. C-H stretch), 1728 (C=O). MALDI MS 2357.3 m/z (MH+) (Theory: 2356.1 m/z (M+)). Elemental Analysis C: 48.32 %; H 5.97 % (Theory C: 47.92 %; H 5.90%). SEC Mw: 3060, Mn: 3000, PDI: 1.02.
Example 18
Synthesis of succinic acid monomethallyl ester (SAME) - 2-Methyl-2-propen-l-ol (4.90 mL, 58.2 mmol) was dissolved in pyridine (20 mL) followed by the addition of succinic anhydride (7.15 g, 71.4 mmol). The reaction mixture was stirred at room temperature for 15 hours before the pyridine was removed under vacuum at 30 0C. The remaining liquid was dissolved in CH2Cl2 (100 mL) and washed two times with cold 0.2 N HCl (100 mL). The organic phase was dried with Na2SO4, gravity filtered, and evaporated to give 9.25 g of a clear liquid (92 % yield). 1H NMR (CDCl3): δ 1.70 (s, 3, CH3), 2.64 (m, 4, -CH2-CH2-),
4.48 (s, 2, -CH2-), 4.88 (m, 1, vinyl CH2), 4.93 (m, 1, vinyl CH2). 13C NMR (CDCl3): δ 178.58 (COOH), 172.05 (COOR), 139.88 (CH), 113.31 (CH2), 68.31 (CH2), 29.11 (CH2), 28.99 (CH2), 19.59 (CH3). FTIR: v (cm-1) 2939 (aliph. C-H stretch), 1711 (C=O). GC-MS 173 m/z (MH+) (Theory: 172 m/z (M+)). Elemental Analysis C: 55.51 %; H 7.09 % (Theory: C: 55.81 %; H 7.02 %).
Example 19
Synthesis of [G2]-PGLSA-SAME - Succinic acid monomethallyl ester (0.826 g, 4.80 mmol), [G2J-PGLSA (0.401 g, 0.170 mmol), and DPTS (0.712 g, 2.42 mmol) were dissolved in THF (50 mL). The reaction flask was flushed with nitrogen and then DCC (1.52 g, 7.37 mmol) was added. Stirring at room temperature was continued for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount Of CH2Cl2 (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 to 5:95 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. The ethyl ether was decanted and the precipitate was isolated to yield 0.558 g of a clear colorless oil (68.2 % yield). 1H NMR (CDCl3): δl.72 (s, 48, CH3), 2.63 (m, 116, -CH2-CH2-), 4.16 (m, 23, -CH2-CH-CH2-), 4.27 (m, 23, -CH2-CH-CH2-), 4.48 (s, 32, -CH2-), 4.89 (s, 16, vinyl CH2), 4.94 (s, 16, vinyl CH2), 5.24 (m, 14, -CH2-CH-CH2-). 13C NMR (CDCl3): δ 171.91 (COOR), 171.67 (COOR), 139.98 (CH), 113.22 (CH2), 69.43 (CH), 68.31 (CH2), 62.56 (CH2), 29.10 (CH2), 29.02 (CH2) 28.83 (CH2), 19.66 (CH3). FTIR: v (χm'1) 2969 (aliph. C-H stretch), 1734 (C=O). MALDI MS 4840.9 m/z (MH+) (Theory: 4838.7 m/z (M+)). Elemental Analysis C: 55.37 %; H 6.22 % (Theory C: 55.35%; H 6.29%). SEC Mw: 5310, Mn: 5230, PDI: 1.02.
Example 20 Synthesis of [G3]-PGLSA-bzld - 2-(c/s-l,3-0-Benzylidene glycerol)succinic acid mono ester (2.77 g, 9.89 mmol), [G2]-PGLSA (1.00 g, 0.425 mmol), and DPTS (1.30 g, 4.42 mmol) were dissolved in THF (40 mL). The reaction flask was flushed with nitrogen and then DCC (2.67 g, 12.9 mmol) was added. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and
washed with a small amount of THF (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 to 5:95 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. The ethyl ether was decanted and the precipitate was isolated to yield 3.51 g of a white powder (90 % yield). 1H NMR (CDCl3): δ 2.57 - 2.72 (broad m, 116, -CH2-CH2-), 4.12 (m, 60, -CH2-CH-CH2-), 4.23 (m, 60, -CH2-CH-CH2-), 4.68 (m, 16, -CH2-CH-CH2-), 5.22 (m, 14, -CH2-CH-CH2-), 5.49 (s, 16, CH), 7.33 (m, 48, arom. CH), 7.46 (m, 32, arom. CH). 13C NMR (CDCl3): δ 172.31 (COOR), 171.97 (COOR), 171.65 (COOR), 138.01 (CH), 129.28 (CH), 128.49 (CH), 126.21 (CH), 101.28 (CH), 69.45 (CH), 69.16 (CH2), 66.53 (CH), 62.59 (CH2), 29.32 (CH2), 29.16 (CH2) 29.01 (CH2), 28.81 (CH2). FTIR: ycm"1) 2984 (aliph. C-H stretch), 1733 (C=O). MALDI MS 6553.4 m/z (MH+) (Theory: 6552.2 m/z (M+)). Elemental Analysis C: 58.50 %; H 5.66 % (Theory C: 58.29 %; H 5.57 %). SEC Mw: 5550, Mn: 5480, PDI: 1.01.
Example 21
Synthesis of [G3] -PGLSA-OH - Pd/C (10 % w/w) was added to a solution of benzylidene protected [G3]-PGLSA (1.23 g, 0.188 mmol) in 9:1 THF/MeOH (20 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 50 psi Of H2 before shaking for 10 hours. The catalyst was filtered and washed with 9:1 THF/MeOH (20 mL). The filtrate was evaporated to give 0.923 g of a colorless, viscous oil (95 % yield). 1H NMR (CD3OD): δ 2.64 (m, 116, -CH2-CH2-), 3.51 (m, 26, -CH2-CH-CH2-), 3.67 (m, 28, -CH2-CH-CH2-), 3.80 (m, 12, -CH2-CH-CH2-), 4.05 (m, 14, -CH2-CH-CH2-), 4.14 (m, 14, -CH2-CH-CH2-), 4.22 (m, 22, -CH2-CH-CH2-), 4.30 (m, 22, -CH2-CH-CH2-), 5.26 (m, 14, -CH2-CH-CH2). 13C NMR (CD3OD): δ 172.86 (COOR), 69.91 (CH), 67.64 (CH), 65.67 (CH2), 62.87 (CH2), 62.41 (CH2), 28.61 (CH2). FTIR: v (cm"1) 3442 (OH), 2959 (aliph. C-H stretch), 1731
(C=O). MALDI MS 5144.8 m/z (MH+) (Theory: 5142.5 m/z (M+)). Elemental Analysis C: 48.07 %; H 5.84 % (Theory C: 48.11 %; H 5.84 %). SEC Mw: 5440, Mn: 5370, PDI: 1.01.
Example 22
Synthesis of [G4]-PGLSA-bzId - 2-(cw-l,3-O-Benzylidene glycerol)succinic acid mono ester (2.43 g, 8.67 mmol), [G3]-PGLSA (0.787 g, 0.153 mmol), and DPTS (1.30 g, 4.42
mmol) were dissolved in 10:1 THF/DMF (40 mL). The reaction flask was flushed with nitrogen and then DCC (2.63 g, 12.7 mmol) was added. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, solvents were removed under vacuum and the remaining solids were redissolved CH2Cl2. The DCC-urea was filtered and washed with a small amount Of CH2Cl2 (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 to 5:95 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. The ethyl ether was decanted and the precipitate was exposed to reduced pressure to yield 1.50 g of a white powder (73 % yield). 1H NMR (CDCl3): δ 2.63 (m, 70, -CH2-CH2-), 2.72 (m, 146, - CH2-CH2-), 2.90 (m, 32, -CH2-CH2-), 4.14 (m, 100, -CH2-CH-CH2-), 4.25 (m, 100, -CH2- CH-CH2-), 4.70 (m, 32, -CH2-CH-CH2-), 5.25 (m, 16, -CH2-CH-CH2-), 5.52 (s, 32, CH), 7.33 (m, 96, arom. CH), 7.47 (m, 64, arom. CH). 13C NMR (CDCl3): δ 172.27 (COOR), 171.90 (COOR), 171.57 (COOR), 138.08 (CH), 129.25 (CH), 128.47 (CH), 126.23 (CH), 101.27 (CH), 69.49 (CH), 69.13 (CH2), 66.54 (CH), 62.45 (CH2), 29.34 (CH2), 29.02
(CH2), 28.83 (CH2). FTIR: v (χm x) 2978 (aliph. C-H stretch), 1733 (C=O). MALDI MS 13536.8 m/z (MH+) (Theory: 13534.7 m/z (M+)). Elemental Analysis C: 58.20 %; H 5.56 % (Theory C: 58.04 %; H 5.56 %). SEC Mw: 9000, Mn: 8900, PDI: 1.01.
Example 23 Synthesis of [G4] -PGLSA-OH - Pd/C (10 % w/w) was added to a solution of benzylidene protected [G4]-PGLSA (0.477 g, 0.0352 mmol) in 9:1 THF/MeOH (20 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 50 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with 9:1 THF/MeOH (20 mL). The filtrate was evaporated to give 0.351 g of a colorless, viscous oil (93 % yield). 1H NMR (CD3OD): δ 2.65 (m, 244, -CH2-CH2-), 3.53 (m, 50, -CH2-CH-CH2), 3.65 (m, 22, -CH2-CH-CH2-), 3.81 (m, 28, -CH2-CH-CH2-), 4.05 (m, 32, -CH2-CH-CH2-), 4.14 (m, 32, -CH2-CH-CH2-), 4.24 (m, 60, -CH2-CH-CH2-), 4.30 (m, 60, -CH2-CH-CH2-), 5.26 (m, 32, -CH2-CH-CH2-). 13C NMR (CD3OD): δ 172.94 (COOR), 69.92 (CH), 65.72 (CH2), 62.91 (CH2), 28.67 (CH2). FTIR: D (cm"1) 3444 (OH), 2931 (aliph. C-H stretch), 1729 (C=O). MALDI MS
10715.6 m/z (MH+) (Theory: 10715.3 m/z (M+)). Elemental Analysis C: 48.50 %; H 5.83 % (Theory C: 48.20 %; H 5.81 %). SEC Mw: 8800, Mn: 8720, PDI: 1.01.
Example 24
The PGLSA dendrimers or other dendrimers described herein can also be synthesized through Accelerated Syntheses for example:
Example 24.1 Synthesis of 2-(cis-l,3-O-benzylidene glycerol)succinic acid mono ester anhydride - 2-(c/s-l,3-(9-Benzylidene glycerol)succinic acid mono ester (50.00 g, 178.4 mmol) ) and DCC (22.09 g, 107.0 mmol) were dissolved in DCM (300 mL) and stirred for 14 hours. The DCU precipitate was collected by filtration and washed with DCM (50 mL). The organic phase was directly added to 900 mL of hexanes. The hexanes and precipitate were cooled to -20 0C for 3 hours before 46.11 g of precipitate was collected after filtration (95 % yield). 1H NMR (CDCl3): δ 2.75 (m, 4, -CH2-CH2-), 4.12 (m, 4, -CH2-CH-CH2-), 4.25 (m, 4, -CH2-CH-CH2-), 4.71 (m, 2, -CH2-CH-CH2-), 5.52 (s, 2, CH), 7.34 (m, 6, arom. CH), 7.47 (m, 4, arom. CH). 13C NMR (CDCl3): δ 171.77 (COOR), 167.99 (-COOCO-), 137.96 (CH), 129.29 (CH), 128.51 (CH), 126.20 (CH), 101.36 (CH), 69.13 (CH2), 66.76 (CH), 30.37 (CH2), 28.94 (CH2). FTIR: v (cm"1) 2938 (aliph. C-H stretch), 1815 (C=O), 1730 (C=O). FAB-MS 543.2 m/z [M-H]+ (Theory: 542.53 m/z [M]+). Elemental Analysis C: 61.83 %; H 5.70 % (Theory: C: 61.99 %; H 5.57 %). Example 24.2 Synthesis of [Gl]-PGLSA-bzld - Pd(OH)2/C (10% w/w) and activated carbon were added to a solution of [GO]-PGLSA-bzld (3.571 g, 8.071 mmol) in THF (25 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst and activated carbon were filtered off and washed with THF (50 mL). 2-(c/s-l,3-Obenzylidene glycerol)succinic acid mono ester anhydride (21.990 g, 40.532 mmol) and then DMAP (0.514 g, 4.207 mmol) were directly added to the deprotected core in the THF ( more THF was added to give a total volume of 100 mL). The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Any remaining anhydride was quenched by the addition of n-propanol (4.0 mL, 44 mmol), which was allowed to stir for another 5 hours. The THF was removed under vacuum and the remaining contents were dissolved in DCM (250 mL) and washed once with 0.1 N HCl (200 mL) and three times with saturated sodium bicarbonate (200 mL). The organic phase
was dried with Na2SO4, filtered, and concentrated before the dendrimer was precipitated in hexanes (450 mL) and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 10.29 g of a white solid (96.9 % yield). 1H NMR, 13C NMR, FTIR, MALDI-TOF MS, Elemental Analysis, and SEC have been previously reported. Tg (0C): 36.7 to 42.4, 39.5 at half-height.
Example 24.3 Synthesis of [G2]-PGLSA-bzld (186) - Pd(OH)2/C (10% w/w) and activated carbon were added to a solution of [Gl]-PGLSA-bzld (4.40 g, 3.43 mmol) in THF (50 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst and activated carbon were filtered off and washed with THF (50 mL). 2-(c/s-l,3-C>-benzylidene glycerol)succinic acid mono ester anhydride (18.459 g, 34.024 mmol) and then DMAP (0.831 g, 6.802 mmol) were directly added to the deprotected dendrimer in the THF. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Any remaining anhydride was quenched by the addition of n-propanol (3.0 mL, 33 mmol), which was allowed to stir for another 5 hours. The THF was removed under vacuum and the remaining contents were dissolved in DCM (400 mL) and washed once with 0.1 N HCl (300 mL) and three times with saturated sodium bicarbonate (300 mL). The organic phase was dried with Na2SO4, filtered, and concentrated before the dendrimer was precipitated in hexanes (900 mL) and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 9.85 g of a white solid (96.2 % yield). 1H NMR, 13C NMR, FTIR, MALDI-TOF MS, Elemental Analysis, and SEC have been previously reported. Tg (0C): 39.3 to 45.4, 42.3 at half-height. Example 24.4 Synthesis of [G3]-PGLSA-bzId - Pd(OH)2/C (10% w/w) and activated carbon were added to a solution of [G2]-PGLSA-bzld (12.81 g, 4.218 mmol) in THF (100 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst and activated carbon were filtered off and washed with THF (100 mL). From this solution, 1.822 g of [G2]-PGLSA-OH in THF was removed form the mixture. Next, 2-(c/s-l,3-0-benzylidene glycerol)succinic acid mono ester anhydride (45.9154 g, 84.632 mmol) and then DMAP (1.5592 g, 12.763 mmol) were directly added to the deprotected core in the THF. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Any remaining anhydride was quenched by the addition of n-propanol (8.0 mL, 88 mmol), which was allowed to stir for
another 5 hours. The THF was removed under vacuum and the remaining contents were dissolved in DCM (500 mL) and washed once with 0.1 N HCl (400 mL) and three times with saturated sodium bicarbonate (400 mL). The organic phase was dried with Na2SO4, filtered, and concentrated before the dendrimer was precipitated in hexanes (800 mL) and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 20.37 g of a white solid (91.4 % yield). 1H NMR, 13C NMR, FTIR, MALDI-TOF MS, Elemental Analysis, and SEC have been previously reported. Tg (0C): 43.1 to 48.3, 45.7 at half-height. Example 24.5 Synthesis of [G3]-PGLSA-OH - Pd(OH)2/C (10% w/w) and activated carbon were added to a solution of [G3]-PGLSA-bzld (3.571 g, 8.071 mmol) in THF/MeOH (9: 1) (25 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst and activated carbon were filtered off and washed with more of the THF/MeOH solution (50 mL) before the solvents were evaporated. The product was used directly in next reaction Example 24.6 Synthesis of [G4]-PGLSA-bzld - The deprotected core was dissolved in the THF/dimethyl acetimide (10:1) (200 mL) and 2-(czs-l,3-0-benzylidene glycerol)succinic acid mono ester anhydride (60.83 g, 0.11212 mmol) and then DMAP (1.63 g, 13.342 mmol) were directly added to the reaction flask. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Any remaining anhydride was quenched by the addition of n-propanol (4.0 mL, 44 mmol), which was allowed to stir for another 5 hours. The solvents were removed under vacuum and the remaining contents were dissolved in DCM (250 mL) and washed once with 0.1 N HCl (200 mL) and three times with saturated sodium bicarbonate (200 mL). The organic phase was dried with Na2SO4, filtered, and concentrated before the dendrimer was precipitated in hexanes (450 mL) and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 33.25 g of a white solid (88.15 % yield). 1H NMR, 13C NMR, FTIR, MALDI-TOF MS, Elemental Analysis, and SEC have been previously reported. Tg (0C): 43.6 to 49.6, 47.0 at half-height. Example 24.7 Synthesis of [G5]-PGLSA-bzld - [G4]-PGLSA-OH (0.2052 g, 0.0192 mmol) and 2-(c/s-l,3-(9-Benzylidene glycerol)succinic acid mono ester anhydride (0.067 g, 0.548 mmol), were dissolved in 1 :1 THF/DMF (15 mL). DMAP (1.152 g, 2.123 mmol) was
added and the reaction flask was flushed with nitrogen. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Any remaining anhydride was quenched by the addition of water (4.0 mL) which was allowed to stir for another 5 hours. The solvents were removed under vacuum and the remaining contents were dissolved in DCM (150 mL) and washed once with 0.1 N HCl (100 mL) and three times with saturated sodium bicarbonate (100 mL). The organic phase was dried with Na2SO4, filtered, and concentrated before the dendrimer was precipitated in hexanes (450 mL) and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 0.414 g of a white solid (78.6 % yield). 1H NMR (CDCl3): δ 2.57-2.69 (broad m, 488, -CH2-CH2-), 4.07-4.21 (m, 507, -CH2-CH-CH2-), 4.66 (m, 64, -CH2-CH-CH2-), 5.19 (m, 63, -CH2-CH- CH2-), 5.48 (s, 64, CH), 7.31 (m, 194, arom. CH), 7.44 (m, 128, arom. CH). 13C NMR (CDCl3): δ 172.28 (COOR), 171.91 (COOR), 171.61 (COOR), 138.08 (CH), 129.25 (CH), 128.47 (CH), 126.23 (CH), 101.24 (CH), 69.47 (CH), 69.12 (CH2), 66.54 (CH), 62.45 (CH2), 29.33 (CH2), 29.17 (CH2), 29.02 (CH2), 28.83 (CH2). MALDI MS 27059 m/z [M- H]+ (Theory: 27500 m/z [M]+). SEC Mw: 16150, Mn: 15870, PDI: 1.02.
Example 25
Syntheses of [Gn]-PGLSA Dendrons with Focal NHS Activated Ester Example 25.1 Synthesis of [2-(cis-l,3-O-benzylidene glycerol)-N-succinimidyl] succinate (bzld-[Gl]-PGLSA-NHS dendron) - 2-(cw-l,3-O-benzylidene glycerol)succinic acid mono ester (11.47 g, 40.92 mmol), N-hydroxy succinimide (4.85 g, 42.18 mmol), and DPTS (4.26 g, 14.50 mmol), were dissolved in CH2Cl2 (100 mL). The reaction flask was flushed with nitrogen and then DCC (13.44 g, 65.14 mmol) was added. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. The DCC-urea was filtered and washed with a small amount of CH2Cl2 (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Following vacuum filtration, 13.0 g of a white solid was collected (84 % yield). 1H NMR (400 MHz, CDCl3): δ 2.76 (broad s, 4, -CH2-CH2-), 2.85 (m, 2, -CH2-CH2-), 2.96 (m, 2, -CH2-CH2-), 4.13 (m, 2, -CH2-CH-CH2-), 4.27 (m, 2, -CH2-CH-CH2-), 4.72 (m, 1, -CH2-CH-CH2-), 5.52 (s, 1, CH), 7.34 (m, 3, arom. CH), 7.47 (m, 2, arom. CH). 13C NMR (400 MHz, CDCl3): δ
171.32 (COOR), 169.12 (COOR), 167.82 (COOR), 137.96 (CH), 129.30 (CH), 128.51 (CH), 126.23 (CH), 101.38 (CH), 69.11 (CH2), 66.94 (CH), 29.08 (CH2), 26.51 (CH2), 25.74 (CH2). FTIR: v (cm"1) 29318 (aliph. C-H stretch), 1820.09 and 1727 (C=O). GC- MS 378 m/z [M-H]+ (Theory: 377 m/z [M]+). Elemental Analysis C: 57.22 %; H 5.07 % (Theory: C: 57.29 %; H 5.08 %).
Example 25.2 Synthesis of bzld-[G2] -PGLSA-NHS dendron - Pd/C (10% w/w) was added to a solution of bzld-[G I]-PGLSA-NHS dendron (0.514 g, 1.36 mmol) in THF (20 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 20 min. The catalyst and activated carbon were filtered off and washed with THF (50 mL). 2-(cw-l,3-O-benzylidene glycerol)succinic acid mono ester (0.975 g, 3.48 mmol) and DPTS (0.475 g, 1.61 mmol) were directly added to this solution. The reaction flask was flushed with nitrogen and then DCC (1.08 g, 5.24 mmol) was added. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. The DCC-urea was filtered and washed with a small amount of THF (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Following vacuum filtration, 0.991 g of a white solid was collected (70 % yield). 1H NMR (400 MHz, CDCl3): δ 2.63 (broad s, 4, -CH2-CH2-), 2.72 (m, 10, -CH2-CH2-), 2.90 (t, 2, -CH2-CH2-), 4.14 (m, 6, -CH2-CH-CH2-), 4.25 (m, 6, -CH2-CH-CH2-), 4.70 (m, 2, -CH2-CH-CH2-), 5.25 (m, 1, -CH2-CH-CH2-), 5.52 (s, 2, CH), 7.33 (m, 6, arom. CH), 7.47 (m, 4, arom. CH). 13C NMR (400 MHz, CDCl3): δ 172.31 (COOR), 171.92 (COOR), 170.35 (COOR), 169.12 (COOR), 167.80 (COOR), 138.02 (CH), 129.27 (CH), 128.49 (CH), 126.21 (CH), 101.33 (CH), 69.97 (CH2), 69.17 (CH2), 66.53 (CH), 62.49 (CH2), 29.38 (CH2), 29.05 (CH2), 26.35 (CH2), 25.74 (CH2). FAB MS 814.3 m/z [M-H]+ (Theory: 813.8 m/z [M]+). Elemental Analysis C: 57.42 %; H 5.40 % (Theory: C: 57.56 %; H 5.33 %).
Example 25.3 Synthesis of bzld-[G3]-PGLSA-NHS dendron - Pd/C (10% w/w) was added to a solution of bzld-[G2]-PGLSA-NHS dendron (0.687 g, 0.844 mmol) in THF (20 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 20 min. The catalyst and activated carbon were filtered off and washed with THF (50 mL). 2-(m-l,3-0-benzylidene glycerol)succinic acid mono ester (1.269 g,
4.53 mmol) and DPTS (0.657 g, 2.23 mmol) were directly added to this solution. The reaction flask was flushed with nitrogen and then DCC (1.08 g, 5.24 mmol) was added. The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. The DCC-urea was filtered and washed with a small amount of THF (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 3:97 methanol: CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. Following vacuum filtration, 0.796 g of a white solid was collected (72 % yield). 1H NMR (400 MHz, CDCl3): δ 2.59 (m, 9, -CH2-CH2-), 2.63 (m, 9, -CH2-CH2-), 2.74 (m, 12, -CH2-CH2-), 2.89 (t, 2, -CH2-CH2-), 4.14 (m, 14, -CH2-CH-CH2-), 4.24 (m, 14, -CH2-CH-CH2-), 4.70 (m, 4, - CH2-CH-CH2-), 5.20 (m, 2, -CH2-CH-CH2-), 5.26 (m, 1, -CH2-CH-CH2-), 5.51 (s, 4, CH), 7.33 (m, 12, arom. CH), 7.47 (m, 8, arom. CH). 13C NMR (400 MHz, CDCl3): δ 172.29 (COOR), 171.92 (COOR), 171.59 (COOR), 169.23 (COOR), 167.87 (COOR), 138.03 (CH), 129.27 (CH), 128.48 (CH), 126.21 (CH), 101.33 (CH), 69.51 (CH2), 69.17 (CH2), 66.54 (CH), 62.51 (CH2), 29.37 (CH2), 29.04 (CH2), 28.86 (CH2), 25.73 (CH2). FAB MS 1686.7 m/z [M-H]+ (Theory: 1686.6 m/z [M]+). Elemental Analysis C: 57.52 %; H 5.53 % (Theory: C: 57.68 %; H 5.44 %).
Example 26 Synthesis of [G2]-PGLSA-(Z)Lys(Z) - Z-Lys(Z)-OH (1.88 g, 4.53 mmol), [G2]-PGLSA (0.401 g, 0.170 mmol), and DPTS (0.66 g, 2.24 mmol) were dissolved in THF (20 mL). The reaction flask was flushed with nitrogen and then DCC (1.43 g, 6.93 mmol) was added. Stirring at room temperature was continued for 24 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount of THF (20 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 2:98 to 4:96 methanol:CH2Cl2. The product was dissolved in CH2Cl2, filtered (to remove any DCU), and precipitated in ethyl ether at -20 0C to remove remaining DCC. The ethyl ether was decanted and the precipitate was isolated to yield 1.69 g of a white solid (95.1 % yield). 1H NMR (CDCl3): δ 1.28 (broad s, 32, -CH2-CH2-CH2- CH2-NH-), 1.43 (broad s, 32, -CH2-CH2-CH2-CH2-NH-), 1.59 (broad s, 16, -CH2-CH2-CH2- CH2-NH-), 1.72 (broad s, 16, -CH2-CH2-CH2-CH2-NH-), 1.59 (broad s, 32, -CH2-CH2-CH2- CH2-NH-), 2.54 (broad s, 52, -CH2-CH2-), 4.09-4.28 (broad m, 23, -CH2-CH-CH2- and -
CH2-CHCO-NH-), 5.00 (s, 32, -CH2-Ph), 5.03 (s, 32, -CH2-Ph), 5.18 (m, 14, -CH2-CH- CH2-), 7.25 (m, 165, arom. CH). 13C NMR (CDCl3): δ 171.98 (COOR), 171.51 (COOR), 156.80 (COOR), 156.34 (COOR), 136.84 (CH), 136.44 (CH), 128.67 (CH), 128.29 (CH), 67.19 (CH), 66.76 (CH), 62.58 (CH2), 53.96 (CH), 40.62 (CH2), 31.80 (CH2) 29.49 (CH2), 28.89 (CH2), 28.73 (CH2), 22.56 (CH2). MALDI MS 8708.0 m/z [M-H]+ (Theory: 8699.0 m/z [M]+). SEC Mw: 7330, Mn: 7220, PDI: 1.01.
Example 27
Synthesis of [G2]-PGLSA-Lys - [G2]-PGLSA-Z-Lys(Z) (59.0 mg, 0.00678 mmol), was dissolved in DMF (3 mL). The reaction flask was flushed with nitrogen and then 10% Pd/C (400 mg) was added and stirred vigorously. To this stirring solution, formic acid was slowly added via syringe. The solution began to bubble and give off heat. Stirring at room temperature was continued for 14 hours under nitrogen atmosphere. Upon completion, Pd/C was filtered and washed with a small amount of 1 N HCl (10 mL), which was added to the DMF solution containing the dendrimer. The resulting solution was added drip wise into a large excess of acetone. The contents were cooled to —20 0C over night. The acetone was decanted and the precipitate was isolated to yield 29.0 mg of product (96.3 % yield). 1H NMR (CDCl3): δ 1.39 (broad m, 32, -CH2-CH2-CH2-CH2-NH-), 1.60 (broad m, 32, -CH2- CH2-CH2-CH2-NH-), 1.83 (broad m, 16, -CH2-CH2-CH2-CH2-NH-), 1.92 (broad m, 16, - CH2-CH2-CH2-CH2-NH-), 2.53-2.60 (broad m, 52, -CH2-CH2-), 2.87 (broad m, 32, -CH2- CH2-CH2-CH2-NH-), 4.08 (broad m, 20, -CH2-CH-CH2- and -CH2-CHCO-NH-), 4.09 (broad m, 23, -CH2-CH-CH2- and -CH2-CHCO-NH-), 4.21 (broad m, 25, -CH2-CH-CH2- and -CH2-CHCO-NH-), 4.35 (broad m, 16, -CH2-CH-CH2- and -CH2-CHCO-NH-), 4.43 (broad m, 16, -CH2-CH-CH2- and -CH2-CHCO-NH-), 5.19 (m, 5, -CH2-CH-CH2-), 5.30 (m, 8, -CH2-CH-CH2-). 13C NMR (CDCl3): δ 174.35 (COOR), 173.74 (COOR), 169.67 (COOR), 70.04 (CH), 64.21 (CH2), 63.01 (CH2), 52.72 (CH2), 39.17 (CH2), 37.07 (CH2), 29.38 (CH2) 28.81 (CH2), 26.44 (CH2), 21.78 (CH2), 21.71 (CH2). MALDI MS 4404 m/z [M-H]+ (Theory: 4407 m/z [M]+). SEC Mw: 7730, Mn: 7580, PDI: 1.02.
Example 28
Synthesis of [G2]-PGLSA-COOH - [G2]-PGLSA-OH (0.636 g, 0.270 mmol) was dissolved in pyridine (20 mL) and stirred while succinic anhydride (0.649 g, 6.485 mmol) was added. The reaction mixture was stirred for 16 hours at 35 oC before the pyridine was
removed under reduced pressure. The contents were partially dissolved in DCM (15 mL), and 0.1 N HCl (15 mL) was then added and the mixture was stirred for an additional 15 minutes. After stirring, the organic and aqueous phases separated and a layer was formed between the two phases. While avoiding the interface, most of the aqueous and organic phases were removed. This washing procedure with 15 mL of DCM and 0.1 N HCl was repeated two more times. Any remaining organic or aqueous phase was removed first by rotoevaporation followed by lyopholization to yield 0.990 g of a highly viscous liquid (92.7 % yield). MALDI MS 3958.4 m/z [M+H]+, (Theory: 3957.2 m/z [M]+).
Example 29 Synthesis of [G4]-PGLSA-COOH and [G4]-PGLSA-COO Na+ - [G4]-PGLSA-OH (0.140 g, 0.0131 mmol) was dissolved in pyridine (10 mL) and stirred while succinic anhydride (0.167 g, 1.68 mmol) was added. The reaction mixture was stirred for 16 hours before the pyridine was removed under reduced pressure. The contents were partially dissolved in DCM (15 mL), and 0.1 N HCl (15 mL) was then added and the mixture was stirred for an additional 15 minutes. After stirring, the organic and aqueous phases separated and a layer was formed between the two phases. While avoiding the interface, most of the aqueous and organic phases were removed. This washing procedure with 15 mL of DCM and 0.1 N HCl was repeated two more times. Any remaining organic or aqueous phase was removed first by rotoevaporation followed by lyopholization to yield 0.191 g of a highly viscous liquid (85 % yield). To dissolve the polymer in water, deionized water (10 mL) and brine (0.5 mL) were added to the solution and 0.05 N NaOH was added drop-wise to the stirring solution until the pH remained at 7.0. The dendrimer was purified via dialysis with 7,000 MW cutoff dialysis tubing for 24 hours in DI water. The water was then removed via lyopholization to obtain a white solid. 1H NMR (D2O): δ 2.32 (m, 130, -CH2- CH2-), 2.46 (m, 133, -CH2-CH2-), 2.58 (m, 228, -CH2-CH2-) 4.13-4.21 (m, 240, -CH2-CH- CH2-), 5.18 (m, 62, -CH2-CH-CH2-). 13C NMR (D2O): δ 180.72 (COOH), 175.37 (COOH), 173.52 (COOR), 70.14 (CH), 69.76 (CH), 62.80 (CH2), 34.31 (CH2), 32.10 (CH2), 30.72 (CH2), 29.01 (CH2). FTIR: v (cm"1) 3368 (OH), 2964 (aliph. C-H stretch), 1732 (C=O), 1567 (asym COO" stretch), 1409 (sym COO" stretch), 1149 (C-O stretch). MALDI MS 17168 m/z [M + Na]+, 8602 m/z [M + Na]2+, (Theory: 17120.0 m/z [M]+). SEC Mw: 8330, Mn: 7780, PDI: 1.11.
Example 30
Synthesis of 2-(tert-ButyldiphenyIsiIanyloxy)-succinic acid 4-(2-phenyI-[l,3]dioxan-5- yl) ester - L-Malic acid (2.00 g, 15.0 mmol) was dissolved in pyridine (25 mL) and tert- butylchlorodiphenylsilane (3.9 mL, 15.0 mmol) was added via syringe. The reaction was stirred for 14 hours before the pyridine was removed by vacuum. The remaining residue was dissolved in DCM (100 mL) and washed with 0.2 N HCl (2x 100 mL). The organic layer was dried with Na2SO4, filtered, and evaporated. Crude 2-{tert- butyldiphenylsilanyloxy) succinic acid was subsequently dissolved in a 2:1 mixture of trifluoroacetic anhydride and THF (50 mL) respectively and heated to 50 0C for 2 hours. The solvents were removed by vacuum and the crude mixture was azeotroped with toluene. The crude anhydride was dissolved in pyridine and czs- 1,3-Obenzylideneglycerol (2.7 g, 54.9 mmol) was added before the solution was stirred another 14 hours. The pyridine was removed by vacuum. The remaining residue was dissolved in DCM (100 mL) and washed with 0.2 N HCl (2x 100 mL). The organic layer was dried with Na2SO4, filtered, and evaporated. The crude product was purified by silica gel chromatography, eluting with 79:20: 1 to 59:40: 1 hexane: ethyl acetate: acetic acid. 0.99 g of a viscous clear liquid were isolated following evaporation of solvents (90 % yield) evaporated to give 1.18 g of a clear viscous oil (12.3% yield). 1H NMR (400 MHz, CDCl3): δ 2.67 (s, 9, -CH3), 2.78 (broad m, 2, -CH2-CH-), 3.64 (broad m, 4, -CH2-CH-CH2-), 4.87 (m, 1, -CH2-CH-CH2-) 2.78 (t, 1, - CH2-CH-), 5.50 (s, 1, CH), 7.34 (broad m, 4, arom. CH), 7.48 (broad m, 11, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 177.54 (COOH), 175.91 (COOH), 171.63 (COOR), 138.00 (CH), 136.20 (CH), 136.14 (CH), 132.94 (CH), 130.20 (CH), 129.25 (CH), 128.41 (CH), 127.95 (CH), 127.81 (CH), 126.35 (CH), 101.41 (CH), 69.43 (CH2), 68.78 (CH), 66.91 (CH2), 39.92 (CH), 26.99 (CH3), 20.95 (CH), 19.53 (CH2). FAB-MS 535.2 m/z [M+H]+ (Theory: 534.67 m/z [M]+).
Example 31
Synthesis of [GO]-PGLAA-bzld - Adipic acid (6.474 g, 44.300 mmol), cw-1,3-0- benzylideneglycerol (17.571 g, 97.508 mmol), and DPTS (10.01 g, 34.03 mmol) were dissolved in DCM (120 mL) followed by the addition of DCC (28.260 g, 136.96 mmol).
The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon
reaction completion, the DCC-urea was filtered and washed with a small amount of DCM (50 mL). The crude product was purified by silica gel chromatography, eluting with 2% MeOH in DCM. The appropriate isolated fractions were concentrated, filtered (to remove any DCU), and directly precipitated in hexanes and cooled to -20 0C overnight. Following vacuum filtration, 12.694 g of a white solid was collected (60.8 % yield). 1H NMR (400 MHz, CDCl3): δ 1.72 (s, 4, -CH2-CH2-CH2-CH2-), 2.45 (s, 4, -CH2-CH2-CH2-CH2-), 4.12 (m, 4, -CH2-CH-CH2-), 4.25 (m, 4, -CH2-CH-CH2-), 4.68 (m, 2, -CH2-CH-CH2-), 5.52 (s, 2, CH), 7.34 (m, 6, arom. CH), 7.48 (m, 4, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 173.47 (COOR), 138.01 (CH), 129.27 (CH), 128.50 (CH), 126.22 (CH), 101.43 (CH), 69.30 (CH2), 66.08 (CH), 34.15 (CH2), 24.49 (CH2). FAB 471.2 m/z [M+H]+ (Theory: 470.51 m/z [M]+).
Example 32 Synthesis of [GO]-PGLAA-OH - Pd(OH)2/C (10% w/w) was added to a solution of [GO]- PGLAA-bzld (2.161 g, 4.593 mmol) in THF (30 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF solution (50 mL). The filtrate was evaporated to give 1.303 g of a clear viscous oil (96.4 % yield). 1H NMR (400 MHz, CD3OD): δ 1.64 (m, 4, -CH2-CH2-CH2-CH2-), 2.36 (m, 4, -CH2-CH2-CH2-CH2-), 3.51 (m, 1, -CH2-CH-CH2-), 3.64 (m, 5, -CH2-CH-CH2-), 3.78 (m, 1, -CH2-CH-CH2-), 4.03 (m, 1, -CH2-CH-CH2-), 4.12 (m, 1, -CH2-CH-CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.76 (COOR), 75.43 (CH), 69.91 (CH), 65.33 (CH2), 62.83 (CH2), 60.49 (CH2), 33.52 (CH2), 33.31 (CH2), 24.12 (CH2). FAB MS 295.30 m/z [M+H]+ (Theory: 294.30 m/z [M]+).
Example 33
Synthesis of adipic anhydride - Adipic acid (96.28 g, 0.6588mol) and acetic anhydride (400 mL) were combined and refluxed at 160 0C for four hours. Afterwards, the acetic acid/anhydride was removed under vacuum. Next the depolymerization catalyst, zinc acetate monohydrate, was added along with a distillation apparatus and the heat was slowly increased. After 100 0C, nothing was collected until 200 0C when 68.79 g of a clear colorless liquid was collected (82.5 % yield). 1H NMR (400 MHz, CDCl3): δ 1.91 (m, 4, -
CH2-CH2-CH2-CH2-), 2.67 (m, 4, -CH2-CH2-CH2-CH2-). 13C NMR (100.6 MHz, CDCl3): δ 168.38 (-COOCO-), 34.60 (CH2), 22.37 (CH2). GC-MS 128 m/z [M]+ (Theory: 128.12 m/z [M]+).
Example 34
Synthesis of 2-(c/s-l,3-0-benzylidene glycerol)adipic acid mono ester c/5-l,3-O-benzylideneglycerol (68.74 g, 0.5365 mol) was dissolved in pyridine (150 mL) followed by the addition of adipic anhydride (82.50 g, 0.4578 mol). The reaction mixture was stirred at room temperature for 18 hours before the pyridine was removed under vacuum at 35 0C. The remaining solid was dissolved in DCM (400 mL) and washed two times with 0.2 N HCl (400 mL), or until the aqueous phase remained at pH 1. The organic phase was evaporated and the solid was added to deionized water (300 mL). 1 N NaOH was added until pH 7 was obtained and the product was in the aqueous solution. The aqueous phase was washed with DCM (400 mL), to extract any remaining adipic anhydride, and then readjusted to pH 4. The aqueous phase was subsequently extracted twice with DCM (400 mL), dried with Na2SO4, filtered, and evaporated to afford 67.53 g of a white powder (47.80 % yield). 1H NMR (400 MHz, CDCl3): δ 1.70 (m, 4, -CH2-CH2- CH2-CH2-), 2.35 (m, 2, -CH2-CH2-CH2-CH2-), 2.44 (m, 2, -CH2-CH2-CH2-CH2-), 4.13 (m, 2, -CH2-CH-CH2-), 4.25 (m, 2, -CH2-CH-CH2-), 4.67 (m, 1, -CH2-CH-CH2-), 5.53 (s, 1, CH), 7.33 (m, 3, arom. CH), 7.47 (m, 2, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 178.98 (COOH), 173.48 (COOR), 137.97 (CH), 129.30 (CH), 128.51 (CH), 126.22 (CH), 101.45 (CH), 69.28 (CH2), 66.13 (CH), 34.13 (CH2), 33.71 (CH2), 24.43 (CH2), 24.21 (CH2). FAB MS 309.1 m/z (MH+) (Theory: 308.33 m/z (M+)).
Example 35
Synthesis of [Gl]-PGLAA-bzld - First, 2-(cis-l,3-0-benzylidene glycerol)adipic acid mono ester (7.226 g, 23.434 mmol), [GO]-PGLAA-OH (1.222 g, 4.152 mmol), and DPTS (2.830 g, 9.621 mmol) were dissolved in THF (100 mL) followed by the addition of DCC (4.32 g, 21.0 mmol). The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon reaction completion, the DCC-urea was filtered and washed with a small amount of THF (50 mL). The crude product was purified by silica gel
chromatography, eluting with 1/1 to 4/1 EtOAc :hexanes. The appropriate isolated fractions were concentrated, filtered (to remove any DCU), and directly precipitated in hexanes and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 5.99 g of a sticky solid (99.1 % yield). 1H NMR (400 MHz, CDCl3): δ 1.63 (m, 20, - CH2-CH2-CH2-CH2-), 2.32 (m, 12, -CH2-CH2-CH2-CH2-), 2.43 (m, 8, -CH2-CH2-CH2-CH2- ), 4.10 (m, 12, -CH2-CH-CH2-), 4.25 (m, 12, -CH2-CH-CH2-), 4.68 (m, 4, -CH2-CH-CH2-), 5.21 (m, 2, -CH2-CH-CH2-), 5.51 (s, 4, CH), 7.32 (m, 12, arom. CH), 7.47 (m, 8, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 173.40 (COOR), 172.87 (COOR), 172.55 (COOR), 138.02 (CH), 129.28 (CH), 128.49 (CH), 126.21 (CH), 101.39 (CH), 69.28 (CH2), 66.11 (CH), 62.39 (CH2), 34.08 (CH2), 33.90 (CH2), 33.75 (CH2), 24.37 (CH2). FAB MS 1455.6 m/z [M+H]+ (Theory: 1455.54 m/z [M]+).
Example 36 Synthesis of [Gl]-PGLAA-OH - Pd(OH)2/C (10% w/w) was added to a solution of [Gl]- PGLAA-bzld (4.870 g, 3.346 mmol) in THF (50 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF solution (50 mL). The filtrate was evaporated to give 3.669 g of a clear viscous oil (99.5 % yield). 1H NMR (400 MHz, CD3OD): δ 1.63 (m, 20, -CH2-CH2-CH2-CH2-), 2.36 (m, 20, -CH2-CH2-CH2-CH2-), 3.52 (m, 2, -CH2-CH-CH2-), 3.59-3.69 (broad m, 12, -CH2-CH-CH2-), 3.79 (m, 1, -CH2-CH-CH2-), 4.03 (m, 1, -CH2- CH-CH2-), 4.14 (m, 5, -CH2-CH-CH2-), 4.32 (m, 4, -CH2-CH-CH2-), 5.24 (m, 2, -CH2-CH- CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.64 (COOR), 173.36 (COOR), 172.93 (COOR), 75.42 (CH), 69.93 (CH), 69.47 (CH), 65.36 (CH2), 62.87 (CH2), 62.15 (CH2), 60.50 (CH2), 33.49 (CH2), 33.35 (CH2), 33.20 (CH2), 24.11 (CH2). MALDI-TOF MS 1125.8 m/z [M+Na]+ (Theory: 1103.11 m/z [M]+).
Example 37
Synthesis of [G2]-PGLAA-bzld - 2-(c/s-l,3-0-benzylidene glycerol)adipic acid mono ester (10.012 g, 32.472 mmol), [Gl]-PGLAA-OH (3.397 g, 3.079 mmol), and DPTS (2.508 g, 8.527 mmol) were dissolved in THF (100 mL) followed by the addition of DCC (4.62 g,
22.4 mmol). The reaction was stirred at room temperature for 14 hours under nitrogen
atmosphere. Upon reaction completion, the DCC-urea was filtered and washed with a small amount of THF (50 mL). The crude product was purified by silica gel chromatography, eluting with 2% MeOH in DCM. The appropriate isolated fractions were concentrated, filtered (to remove any DCU), and directly precipitated in hexanes and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 9.39 g of a sticky wax (89.0 % yield). 1H NMR (400 MHz, CDCl3): δ 1.63 (m, 52, -CH2-CH2-CH2- CH2-), 2.31 (m, 36, -CH2-CH2-CH2-CH2-), 2.41 (m, 16, -CH2-CH2-CH2-CH2-), 4.05 (m, 28, -CH2-CH-CH2-), 4.25 (m, 28, -CS2-CH-CH2-), 4.67 (m, 8, -CH2-CH-CH2-), 5.21 (m, 6, - CH2-CH-CH2-), 5.51 (s, 8, CH), 7.33 (m, 24, arom. CH), 7.46 (m, 16, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 173.39 (COOR), 172.87 (COOR), 172.54 (COOR), 138.02 (CH), 129.27 (CH), 128.49 (CH), 126.21 (CH), 101.38 (CH), 69.27 (CH2), 66.11 (CH), 62.39 (CH2), 34.08 (CH2), 33.74 (CH2), 33.67 (CH2), 24.37 (CH2). MALDI MS 3449.2 m/z [M+Na]+ (Theory: 3425.61 m/z [M]+).
Example 38
Synthesis of [G2] -PGLAA-OH - Pd(OH)2/C (10% w/w) was added to a solution of [G2]- PGLAA-bzld (8.02 g, 2.34 mmol) in THF (100 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF solution (50 mL). The filtrate was evaporated to give 6.360 g of a clear viscous oil (99.4 % yield). 1H NMR (400 MHz, CD3OD): δ 1.62 (m, 52, -CH2- CH2-CH2-CH2-), 2.35 (m, 52, -CH2-CH2-CH2-CH2-), 3.52 (m, 5, -CH2-CH-CH2-), 3.59- 3.71 (broad m, 25, -CH2-CH-CH2-), 3.79 (m, 3, -CH2-CH-CH2-), 4.03 (m, 3, -CH2-CH- CH2-), 4.14 (m, 15, -CH2-CH-CH2-), 4.33 (m, 12, -CH2-CH-CH2-), 5.25 (m, 6, -CH2-CH- CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.63 (COOR), 173.27 (COOR), 172.92 (COOR), 75.42 (CH), 69.94 (CH), 69.47 (CH), 65.38 (CH2), 62.89 (CH2), 62.17 (CH2), 60.52 (CH2), 33.51 (CH2), 33.39 (CH2), 33.22 (CH2), 24.12 (CH2). MALDI-TOF MS 2744.3 m/z [M+Na]+ (Theory: 2720.75 m/z [M]+).
Example 39 Synthesis of [G3]-PGLAA-bzld - 2-(c/s-l,3-0-benzylidene glycerol)adipic acid mono ester (12.626 g, 40.950 mmol), [G2]-PGLAA-OH (5.263 g, 1.934 mmol), and DPTS (3.232
g, 10.989 mmol) were dissolved in THF (100 mL) followed by the addition of DCC (12.581 g, 60.975 mmol). The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon reaction completion, the DCC-urea was filtered and washed with a small amount of THF (60 mL). The crude product was purified by silica gel chromatography, eluting with 1.5 to 3.0 % MeOH in DCM. The appropriate isolated fractions were concentrated, filtered (to remove any DCU), and directly precipitated in hexanes and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 12.22 g of a sticky wax (85.8 % yield). 1H NMR (400 MHz, CDCl3): δ 1.63 (broad m, 130, -CH2-CH2-CH2-CH2-), 2.31 (m, 90, -CH2-CH2-CH2-CH2-), 2.41 (m, 32, -CH2-CH2- CH2-CH2-), 4.10 (m, 62, -CH2-CH-CH2-), 4.24 (m, 62, -CH2-CH-CH2-), 4.67 (m, 16, -CH2- CH-CH2-), 5.19 (m, 14, -CH2-CH-CH2-), 5.51 (s, 16, CH), 7.32 (m, 48, arom. CH), 7.46 (m, 32, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 173.38 (COOR), 172.89 (COOR), 172.48 (COOR), 138.03 (CH), 129.27 (CH), 128.49 (CH), 126.21 (CH), 101.36 (CH), 69.26 (CH2), 66.11 (CH), 62.29 (CH2), 34.08 (CH2), 33.83 (CH2), 33.74 (CH2), 33.67 (CH2), 24.43 (CH2), 24.36 (CH2). MALDI-TOF MS 7390 m/z [M+Na]+ (Theory: 7365.73 m/z [M]+).
Example 40 Synthesis of [G3]-PGLAA-OH - Pd(OH)2/C (10% w/w) was added to a solution of [G3]- PGLAA-bzld (11.03 g, 1.497 mmol) in THF (125 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF solution (75 mL). The filtrate was evaporated to give 8.69 g of a clear viscous oil (97.5 % yield). 1H NMR (400 MHz, CD3OD): δ 1.63 (m, 124, -CH2-CH2-CH2-CH2-), 2.35 (m, 127, -CH2-CH2-CH2-CH2-), 3.52 (m, 7, -CH2-CH- CH2-), 3.60-3.71 (broad m, 55, -CH2-CH-CH2-), 3.79 (m, 4, -CH2-CH-CH2-), 4.04 (m, 5, - CH2-CH-CH2-), 4.14 (m, 34, -CH2-CH-CH2-), 4.32 (m, 29, -CH2-CH-CH2-), 5.25 (m, 14, - CH2-CH-CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.82 (COOR), 173.63 (COOR), 173.36 (COOR), 173.27 (COOR), 172.92 (COOR), 75.45 (CH), 75.40 (CH), 69.96 (CH), 69.48 (CH), 65.40 (CH2), 62.92 (CH2), 62.23 (CH2), 60.54 (CH2), 33.53 (CH2), 33.25 (CH2), 24.15 (CH2). MALDI-TOF MS 5975.0 m/z [M+Na]+ (Theory: 5956.02 m/z [M]+).
Example 41
Synthesis of [GO]-PGLSA-[Gl]-PGLAA-bzld - 2-(cw-l,3-C>-benzylidene glycerol)adipic acid mono ester (11.793 g, 38.248 mmol), [GO]-PGLSA-OH (1.185 g, 4.449 mmol), and DPTS (2.853 g, 9.700 mmol) were dissolved in THF (50 mL) followed by the addition of DCC (7.216 g, 34.973 mmol). The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount of THF (50 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 1/1 to 4/1 EtOAc:hexanes. The appropriate isolated fractions were concentrated, filtered (to remove any remaining DCU), and directly precipitated in hexanes and cooled to —20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 7.173 g of a sticky solid (97 % yield). 1H NMR (400 MHz, CDCl3): δ 1.65 (m, 16, -CH2-CH2-CH2-CH2-), 2.33 (m, 8, -CH2-CH2- CH2-CH2-), 2.42 (m, 8, -CH2-CH2-CH2-CH2-), 2.59 (m, 4, -CH2-CH2-), 4.11 (m, 12, -CH2- CH-C]H2-), 4.24 (m, 12, -CH2-CH-CH2-), 4.67 (m, 4, -CH2-CH-CH2-), 5.20 (m, 2, -CH2- CH-CH2-), 5.51 (s, 4, CH), 7.33 (m, 12, arom. CHJ, 7.47 (m, 8, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 173.41 (COOR), 172.92 (COOR), 171.48 (COOR), 138.02 (CH), 129.28 (CH), 128.49 (CH), 126.21 (CH), 101.38 (CH), 69.65 (CH), 69.27 (CH2), 66.11 (CH), 62.19 (CH2), 34.09 (CH2), 33.73 (CH2), 28.97 (CH2), 24.44 (CH2), 24.36 (CH2). FAB MS 1425.5 m/z [M+H]+ (Theory: 1427.49 m/z [M]+). SEC Mw: 1670, Mn: 1650, PDI: 1.01.
Example 42
Synthesis of [G0]-PGLSA-[G1]-PGLAA-OH - Pd(OH)2/C (10% w/w) was added to a solution of [G0]-PGLSA-[Gl]-PGLAA-bzld (5.900 g, 4.133 mmol) in THF (50 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF (50 mL). The filtrate was evaporated to give 4.407 g of a colorless, viscous oil (99 % yield). 1H NMR (400 MHz, CD3OD): δ 1.63 (m, 16, -CH2-CH2-CH2-CH2-), 2.36 (m, 16, -CH2-CH2-CH2-CH2-), 2.61 (m, 4, -CH2-CH2-), 3.52 (m, 3, -CH2-CH-CH2-), 3.59-3.65 (broad m, 9, -CH2-CH-CH2-), 3.69 (m, 2, -CH2-CH-CH2-), 3.79 (m, 2, -CH2-CH-CH2-), 4.03 (m, 2, -CH2-CH-CH2-), 4.15 (m, 5, -CH2-CH-CH2-), 4.30 (m, 4, -CH2-CH-CH2-), 5.25 (m, 2, -CH2-CH-CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.85 (COOR), 173.67 (COOR), 173.41 (COOR), 171.95
(COOR), 75.42 (CH), 69.93 (CH), 69.78 (CH), 65.36 (CH2), 62.87 (CH2), 62.04 (CH2), 60.50 (CH2), 33.50 (CH2), 33.29 (CH2), 33.19 (CH2), 28.61 (CH2), 24.12 (CH2). MALDI- TOF MS 1097.5 m/z [M+Na]+ (Theory: 1075.06 m/z [M]+). SEC Mw: 1680, Mn: 1660, PDI: 1.01.
Example 43
Synthesis of [G0]-PGLSA-[Gl]-PGLAA-[G2]-PGLSA-bzld - 2-(c/s-l,3-O-benzylidene glycerol)succinic acid mono ester (12.758 g, 45.520 mmol), [GO]-PGLSA-[G1]-PGLAA- OH (4.284 g, 3.984 mmol), and DPTS (5.112 g, 17.381 mmol) were dissolved in THF (100 mL) followed by the addition of DCC (13.912 g, 67.436 mmol). The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount of THF (50 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 2% MeOH in DCM. The appropriate isolated fractions were concentrated, filtered (to remove any remaining DCU), and directly precipitated in hexanes and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 10.84 g of a white solid (85.7 % yield). 1H NMR (400 MHz, CDCl3): δ 1.60 (m, 17, -CH2-CH2-CH2-CH2-), 2.30 (m, 17, -CH2-CH2-CH2-CH2-), 2.63 (m, 20, -CH2-CH2-), 2.72 (m, 16, -CH2-CH2-), 4.11 (m, 29, -CH2-CH-CH2-), 4.23 (m, 29, -CH2-CH-CH2-), 4.70 (m, 8, -CH2-CH-CH2-), 5.20 (m, 6, - CH2-CH-CH2-), 5.51 (s, 8, CH), 7.34 (m, 12, arom. CH), 7.46 (m, 8, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 173.41 (COOR), 172.92 (COOR), 171.48 (COOR), 138.02 (CH), 129.28 (CH), 128.49 (CH), 126.21 (CH), 101.38 (CH), 69.65 (CH), 69.27 (CH2), 66.11 (CH), 62.19 (CH2), 34.09 (CH2), 33.73 (CH2), 28.97 (CH2), 24.44 (CH2), 24.36 (CH2). MALDI-TOF MS 3172.7 m/z [M+Na]+ (Theory: 3173.13 m/z [M]+). SEC Mw: 3600, Mn: 3540, PDI: 1.02.
Example 44
Synthesis of [G0]-PGLSA-[G1]-PGLAA-[G2]-PGLSA-OH - Pd(OH)2/C (10% w/w) was added to a solution of [G0]-PGLSA-[Gl]-PGLAA-[G2]-PGLSA-bzld (5.251 g, 1.655 mmol) in THF (100 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with
THF (50 mL). The filtrate was evaporated to give 4.011 g of a colorless, viscous oil (98.2 % yield). 1H NMR (400 MHz, CD3OD): δ 1.62 (m, 17, -CH2-CH2-CH2-CH2-), 2.36 (m, 17, -CH2-CH2-CH2-CH2-), 2.64 (m, 36, -CH2-CH2-), 3.52 (m, 2, -CH2-CH-CH2-), 3.60-3.66 (broad m, 26, -CH2-CH-CH2-), 3.69 (m, 9, -CH2-CH-CH2-), 3.80 (m, 1, -CH2-CH-CH2-), 4.18 (m, 14, -CH2-CH-CH2-), 4.32 (m, 12, -CH2-CH-CH2-), 5.25 (m, 6, -CH2-CH-CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.38 (COOR), 173.05 (COOR), 172.56 (COOR), 172.24 (COOR), 172.00 (COOR), 75.81 (CH), 69.80 (CH), 69.35 (CH), 67.65 (CH2), 65.68 (CH2), 62.87 (CH2), 62.42 (CH2), 62.11 (CH2), 60.43 (CH2), 33.49 (CH2), 33.20 (CH2), 28.83 (CH2), 28.64 (CH2), 25.28 (CH2), 24.09 (CH2). MALDI-TOF MS 2492.0 m/z [M+Na]+ (Theory: 2468.27 m/z [M]+). SEC Mw: 3390, Mn: 3340, PDI: 1.02.
Example 45
Synthesis of [GO]-PGLSA-[Gl]-PGLAA-[GZ]-PGLSA-[GS]-PGLAA^zId - 2-(c/s-l,3- O-benzylidene glycerol)adipic acid mono ester (10.751 g, 34.869 mmol), [GO]-PGLSA- [G1]-PGLAA-[G2]-PGLSA-OH (3.771 g, 1.528 mmol), and DPTS (1.463 g, 4.975 mmol) were dissolved in THF (120 mL) followed by the addition of DCC (10.598 g, 51.365 mmol). The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount of THF (50 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 1.5% MeOH in DCM. The appropriate isolated fractions were concentrated, filtered (to remove any remaining DCU), and directly precipitated in hexanes and cooled to -20 0C overnight. The hexanes were decanted and the precipitate was isolated to yield 9.88 g of a sticky solid (90.9 % yield). 1H NMR (400 MHz, CDCl3): δ 1.65 (m, 81, -CH2-CH2-CH2-CH2-), 2.31 (m, 52, -CH2-CH2-CH2-CH2-), 2.42 (m, 32, -CH2- CH2-CH2-CH2-), 2.58 (m, 36 -CH2-CH2-), 4.10 (m, 62, -CH2-CH-CH2-), 4.23 (m, 62, -CH2- CH-CH2-), 4.66 (m, 16, -CH2-CH-CH2-), 5.19 (m, 14, -CH2-CH-CH2-), 5.51 (s, 16, CH), 7.33 (m, 47, arom. CH), 7.46 (m, 32, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 173.39 (COOR), 172.90 (COOR), 171.82 (COOR), 171.53 (COOR), 138.04 (CH), 129.26 (CH), 128.49 (CH), 126.22 (CH), 101.36 (CH), 69.65 (CH), 69.26 (CH2), 66.11 (CH), 62.64 (CH2), 62.15 (CH2), 34.07 (CH2), 33.73 (CH2), 28.96 (CH2), 28.80 (CH2), 24.43 (CH2),
24.35 (CH2). MALDI-TOF MS 7137.3 m/z [M+Na]+ (Theory: 7113.25 m/z [M]+). SEC Mw: 7160, Mn: 7060, PDI: 1.01.
Example 46 Synthesis of [GO]-PGLSA-[Gl]-PGLAA-[GZ]-PGLSA-[Ga]-PGLAA-OH - Pd(OH)2/C (10% w/w) was added to a solution of [GO]-PGLSA-[G1]-PGLAA-[G2]-PGLSA-[G3]- PGLAA-bzld (9.175 g, 1.290 mmol) in THF (100 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi Of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF (50 mL). The filtrate was evaporated to give 7.218 g of a colorless, viscous oil (98.1 % yield). 1H NMR (400 MHz, CD3OD): δ 1.63 (m, 83, -CH2-CH2-CH2-CH2-), 2.37 (m, 83, -CH2-CH2-CH2-CH2-), 2.61 (m, 36, -CH2-CH2-), 3.52 (m, 8, -CH2-CH-CH2-), 3.60-3.71 (broad m, 57, -CH2-CH-CH2-), 3.80 (m, 4, -CH2- CH-CH2-), 4.03 (m, 5, -CH2-CH-CH2-), 4.11-4.23 (m, 34, -CH2-CH-CH2-), 4.30 (m, 29, - CH2-CH-CH2-), 5.25 (m, 14, -CH2-CH-CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.85 (COOR), 173.67 (COOR), 173.41 (COOR), 171.95 (COOR), 75.42 (CH), 69.93 (CH), 69.78 (CH), 65.36 (CH2), 62.87 (CH2), 62.04 (CH2), 60.50 (CH2), 33.50 (CH2), 33.29 (CH2), 33.19 (CH2), 28.61 (CH2), 24.12 (CH2). MALDI-TOF MS 5730.3 m/z [MH-Na]+ (Theory: 5703.54 m/z [M]+). SEC Mw: 6570, Mn: 6490, PDI: 1.01.
Example 47
Synthesis of [GO]-PGLSA-[G1]-PGLAA-[G2]-PGLSA-[G3]-PGLAA-[G4]-PGLSA- bzld - 2-(c/s-l,3-0-benzylidene glycerol)succinic acid mono ester (11.572 g, 41.286 mmol), [GO]-PGLSA-[G 1]-PGLAA-[G2]-PGLSA-[G3]-PGLAA-OH (5.593 g, 0.981 mmol), and DPTS (4.094 g, 13.919 mmol) were dissolved in THF (80 mL) followed by the addition of DCC (12.596 g, 61.048 mmol). The reaction was stirred at room temperature for 14 hours under nitrogen atmosphere. Upon completion, the DCC-urea was filtered and washed with a small amount of THF (50 mL) and the solvent was evaporated. The crude product was purified by silica gel chromatography, eluting with 1.5% to 5.0% MeOH in DCM. The appropriate isolated fractions were concentrated, filtered (to remove any remaining DCU), and directly precipitated in hexanes and cooled to -20 0C over 48 hours. The hexanes were decanted and the precipitate was isolated to yield 11.50 g of a white solid
(83.2 % yield). 1H NMR (400 MHz, CDCl3): δ 1.59 (m, 83, -CH2-CH2-CH2-CH2-), 2.30 (m, 83, -CH2-CH2-CH2-CH2-), 2.62 (m, 104, -CH2-CH2-), 2.70 (m, 63, -CH2-CH2-), 4.12 (m, 130, -CH2-CH-CH2-), 4.22 (m, 130, -CH2-CH-CH2-), 4.68 (m, 32, -CH2-CH-CH2-), 5.18 (m, 30, -CH2-CH-CH2-), 5.50 (s, 32, CH), 7.33 (m, 97, arom. CH), 7.46 (m, 66, arom. CH). 13C NMR (100.6 MHz, CDCl3): δ 172.88 (COOR), 172.53 (COOR), 172.25 (COOR), 171.89 (COOR), 138.04 (CH), 129.26 (CH), 128.48 (CH), 126.22 (CH), 101.28 (CH), 69.14 (CH2), 66.54 (CH), 62.60 (CH2), 33.81 (CH2), 33.66 (CH2), 29.35 (CH2), 29.03 (CH2), 24.30 (CH2). SEC Mw: 10440, Mn: 10290, PDI: 1.02.
Example 48 Synthesis of [GO]-PGLSA-[G1]-PGLAA-[G2]-PGLSA-[G3]-PGLAA-[G4]-PGLSA-
OH - Pd(OH)2/C (10% w/w) was added to a solution of [GO]-PGLSA-[G 1] -PGLAA- [G2]- PGLSA-[G3]-PGLAA-[G4]-PGLSA-bzld (2.084 g, 0.1478 mmol) in THF (80 mL). The flask for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 10 hours. The catalyst was filtered and washed with THF (75 mL). The filtrate was evaporated to give 1.652 g of a colorless, viscous oil (99.1 % yield). 1H NMR (400 MHz, CD3OD): δ 1.62 (m, 80, -CH2-CH2-CH2-CH2-), 2.37 (m, 80, -CH2-CH2-CH2-CH2-), 2.64 (m, 164, -CH2-CH2-), 3.52 (m, 12, -CH2-CH-CH2-), 3.63-3.71 (broad m, 160, -CH2-CH- CH2-), 3.80 (m, 6, -CH2-CH-CH2-), 4.06 (m, 14, -CH2-CH-CH2-), 4.20 (m, 62, -CH2-CH- CH2-), 4.30 (m, 60, -CH2-CH-CH2-), 5.25 (m, 30, -CH2-CH-CH2-). 13C NMR (100.6 MHz, CD3OD): δ 173.40 (COOR), 173.06 (COOR), 172.58 (COOR), 75.82 (CH), 69.90 (CH), 69.34 (CH), 67.64 (CH2), 62.45 (CH2), 62.15 (CH2), 60.46 (CH2), 33.25 (CH2), 28.87 (CH2), 28.67 (CH2), 25.27 (CH2), 24.12 (CH2). MALDI-TOF MS 11299.1 m/z [M+Na]+ (Theory: 11276.39 m/z [M]+). SEC Mw: 9150, Mn: 9000, PDI: 1.02.
Example 49
Synthesis of PEG-([G0]-PGLSA-bzld)2 - This example is shown for PEG of 3400 Mw, but we have also used PEG of 10,000 and 20,000 Mw. PEG, Mn=3400, (10.0 g, 2.94 mmol), which was dried under vacuum at 120 0C for three hours, and [2-(cis-l ,3-0- benzylidene glycerol)-N-succinimidyl] succinate (4.03 g, 10.7 mmol) were dissolved in CH2Cl2 (100 mL) and stirred under nitrogen. TEA (2.0 mL, 14 mmol) was added by syringe
and stirring was continued for 14 hours. Any remaining activated ester was quenched by the addition of fresh TEA (1.0 mL, 7.2 mmol) and n-propanol (1.0 mL, 11 mmol), which was allowed to stir for another 10 hours. After removing most of the solvent, the product was precipitated in cold ethyl ether (700 mL) and collected to yield 11.1 g of a white solid (97 % yield). 1H NMR obtained. Elemental Analysis C: 55.31 %; H 8.58 % (Theory C: 55.56 %; H 8.66 %). MALDI MS Mw: 4020, Mn: 3940, PDI: 1.02. SEC Mw: 3980, Mn: 3950, PDI: 1.03.
Example 50 Synthesis of PEG-([G0]-PGLSA-OH)2 - Pd/C (10 % w/w) was added to a solution of PEG-([G0]-PGLSA-bzld)2 (5.07 g, 1.29 mmol) in 80 mL of 9:1 ethyl acetate/methanol. The apparatus for catalytic hydrogenolysis was evacuated and filled with 50 psi of H2 before shaking for 8 hours. The catalyst was filtered off and washed with ethyl acetate (20 mL). The filtrate was evaporated and the remaining white solid was redissolved in a minimal amount Of CH2Cl2 (15 mL)and precipitated in cold ethyl ether (600 mL) to give 4.52 g of a white solid (93 % yield). 1H NMR obtained. Elemental Analysis C: 53.49 %; H 8.78 % (Theory C: 53.69 %; H 8.85 %). MALDI MS Mw: 3780, Mn: 3730, PDI: 1.01. SEC Mw: 3860, Mn: 3710, PDI: 1.021.
Example 51 Synthesis of PEG-([Gl]-PGLSA-bzld)2 - PEG-([G0]-PGLSA-OH)2 (5.81 g, 1.55 mmol), which was dried under vacuum at 80 0C for three hours, and [2-(cώ-l,3-O-benzylidene glycerol)-N-succinimidyl] succinate (4.35 g, 11.5 mmol) were dissolved in CH2Cl2 (70 mL) and stirred under nitrogen. TEA (1.75 mL, 13.0 mmol) was added by syringe and stirring was continued for 14 hours. Any remaining activated ester was quenched by the addition of fresh TEA (1.0 mL, 7.2 mmol) and n-propanol (1.0 mL, 11 mmol), which was allowed to stir for another 10 hours. After removing most of the solvent, the product was precipitated in cold ethyl ether (700 mL) and collected to yield 7.15 g (96 % yield). 1H NMR obtained. MALDI MS Mw: 4520, Mn: 4480, PDI: 1.01. SEC Mw: 4420, Mn: 4240, PDI: 1.04.
Example 52
Synthesis of PEG-([G1]-PGLSA-OH)2 - Pd/C (10 % w/w) was added to a solution of PEG-([Gl]-PGLSA-bzld)2 (5.53 g, 1.15 mmol) in 80 mL of 9:1 ethyl acetate/methanol. The apparatus for catalytic hydrogenolysis was evacuated and filled with 50 psi of H2 before shaking for 8 hours. The catalyst was filtered off and washed with ethyl acetate (20 mL). The filtrate was evaporated and the remaining white solid was redissolved in a minimal amount Of CH2Cl2 (15 mL) and precipitated in cold ethyl ether (700 mL) to give 4.71 g of a white solid (92 % yield). 1H NMR obtained. MALDI MS Mw: 4320, Mn: 4280, PDI: 1.01. SEC Mw: 4390, Mn: 4230, PDI: 1.04.
Example 53
Synthesis of PEG-([G1]-PGLSA-MA)2 - PEG-([G1]-PGLSA-OH)2 (1.03 g, 0.232 mmol), which was dried under vacuum at 80 0C for three hours, was dissolved in CH2Cl2 (40 mL) and stirred under nitrogen before the addition of methacryloyl chloride (1.93 g, 5.12 mmol). TEA (0.80 mL, 5.74 mmol) was added by syringe and stirring was continued for 14 hours. The mixture was diluted with more CH2Cl2 (60 mL) and washed twice with 0.1 N HCl (100 mL). After drying with Na2SO4, filtering, and removing most of the solvent, the product was precipitated in cold ethyl ether and collected to yield 1.08 g (94 % yield). 1H NMR obtained. SEC Mw: 4610, Mn: 4420, PDI: 1.04.
Example 54 Synthesis of PEG-([G2]-PGLSA-bzld)2 - PEG-([G I]-PGLSA-OH)2 (0.697 g, 0.150 mmol), which was dried under vacuum at 80 0C for three hours, and [2-(cis-l ,3-0- benzylidene glyceroty-N-succinimidyl] succinate (1.01 g, 2.68 mmol) were dissolved in CH2Cl2 (30 mL) and stirred under nitrogen. TEA (0.50 mL, 3.59 mmol) was added by syringe and stirring was continued for 14 hours. Any remaining activated ester was quenched by the addition of fresh TEA (1.0 mL, 7.2 mmol) and n-propanol (1.0 mL, 11 mmol), which was allowed to stir for another 10 hours. After removing most of the solvent, the product was precipitated in cold ethyl ether (400 mL) and collected to yield 0.940 g (93 % yield). 1H NMR obtained.
Example 55
Synthesis of ([GI]-PGLSA-MA)2-PEG - ([GI]-PGLSA-OH)2-PEG (0.500 g, 0.113 mmol) was dissolved in DCM (15 mL) and stirred under nitrogen before methacrylic anhydride (0.56 mL, 3.76 mmol) was added by syringe. DMAP (86.0 mg, 0.704 mmol) was added and stirring was continued for 14 hours. Any remaining anhydride was quenched by the addition of methanol (0.1 mL, 3.95 mmol), which was allowed to stir for another 5 hours. The reaction was diluted with DCM (35 mL) and washed with 0.1 N HCl (50 mL) and brine (50 mL). The organic phase was dried with Na2SO4 and filtered before the PEG-based dendrimer was precipitated in cold (-20 0C) ethyl ether (300 mL) and collected to yield 0.519 g of a white solid (93 % yield). 1H NMR (CDCl3): δ 1.90 (m, 19, -CH3), 2.61 (m, 21, -CH2-CH2-), 3.42 (t, 2, -CH2-CH2-), 3.55-3.65 (broad m, 285, -CH2-CH2-), 3.77 (t, 2, -CH2- CH2-), 4.09-4.37 (broad m, 29, -CH2-CH-CH2-), 5.22 (m, 2, -CH2-CH-CH2-), 5.35 (m, 2, - CH2-CH-CH2-), 5.57 (m, 6, CH), 6.07 (m, 6, CH). 13C NMR (CDCl3): δ 171.89 (COOR), 135.84 (CH), 126.64 (CH), 70.75 (CH2), 69.45 (CH), 62.61 (CH2), 28.87 (CH2), 18.43 (CH3). FTIR: v (cnT1) 2873 (aliph. C-H stretch), 1736 (C=O). MALDI MS Mw: 5012, Mn: 4897, PDI: 1.02. SEC Mw: 3910, Mn: 3740, PDI: 1.04. Tm = 40.8.
Example 56
Synthesis of ([G2]-PGLSA-bzld)2-PEG - ([G1]-PGLSA-OH)2-PEG (3.25 g, 0.737 mmol), and 2-(cw-l,3-<9-benzylidene glycerol)succinic acid mono ester anhydride (12.68 g, 23.37 mmol) were dissolved in DCM (50 mL) and stirred under nitrogen. DMAP (0.588 g, 4.81 mmol) was added and stirring was continued for 14 hours. Any remaining anhydride was quenched by the addition of n-propanol (2.5 mL, 28 mmol), which was allowed to stir for another 5 hours. The reaction was diluted with DCM (50 mL) and washed with 0.1 N HCl (100 mL), saturated sodium bicarbonate (100 mL 3x), and brine (100 mL). The organic phase was dried with Na2SO4, filtered, and concentrated before the PEG-based dendrimer was precipitated in cold (-20 0C) ethyl ether (400 mL) and collected to yield 4.57 g of a white solid (91 % yield). 1H NMR (CDCl3): δ 2.61 (broad m, 40, -CH2-CH2-), 2.72 (broad m, 16, -CH2-CH2-), 3.43 (t, 2, -CH2-CH2-), 3.55-3.65 (broad m, 280, -CH2-CH2-), 3.77 (t, 2, -CH2-CH2-), 4.13 (broad m, 28, -CH2-CH-CH2-), 4.22 (broad m, 28, -CH2-CH- CH2-), 4.69 (m, 8, -CH2-CH-CH2-), 5.20 (m, 6, -CH2-CH-CH2-), 5.50 (s, 8, CH), 7.32 (m,
24, arom. CH), 7.46 (m, 16, arom. CH). 13C NMR (CDCl3): δ 172.28 (COOR), 171.91 (COOR), 171.57 (COOR), 138.01 (CH), 129.26 (CH), 128.48 (CH), 126.21 (CH), 101.33 (CH), 70.56 (CH2), 69.50 (CH), 69.16 (CH2), 66.53 (CH), 64.08 (CH2), 29.49 (CH2), 29.21 (CH2). FTIR: v (cm 1) 2879(aliph. C-H stretch), 1736 (C=O). MALDI MS Mw: 6642, Mn: 6492, PDI: 1.02. SEC Mw: 4860, Mn: 4680, PDI: 1.04. Tm = 31.4.
Example 57
Synthesis of ([G2]-PGLSA-OH)2-PEG - Pd(OH)2/C (10 % w/w) was added to a solution of ([G2]-PGLSA-bzld)2-PEG (3.26 g, 0.500 mmol) in 25 mL of 2:1 DCM/methanol. The apparatus for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 8 hours. The catalyst was filtered off and washed with DCM (20 mL). The PEG-based dendrimer was isolated after evaporation of solvents to give 2.86 g of a white solid (98 % yield). 1H NMR (CDCl3): δ 2.63 (broad m, 56, -CH2-CH2-), 3.42 (s, 4, -CH2- CH2-), 3.50-3.67 (broad m, 285, -CH2-CH2-), 3.72 (broad m, 27, -CH2-CH-CH2-), 4.14-4.29 (broad m, 32, -CH2-CH-CH2-), 4.88 (m, 8, -CH2-CH-CH2-), 5.22 (m, 6, -CH2-CH-CH2-). 13C NMR (CDCl3): δ 172.56 (COOR), 172.32 (COOR), 76.01 (CH), 70.78 (CH2), 69.56 (CH), 69.22 (CH2), 64.14 (CH2), 63.52 (CH2), 62.60 (CH2), 61.93 (CH2), 29.44 (CH2), 29.21 (CH2), 28.98 (CH2). FTIR: v (cm"1) 3452 (OH), 288. (aliph. C-H stretch), 1735 (C=O). MALDI MS Mw: 5910, Mn: 5788, PDI: 1.02. SEC Mw: 5340, Mn: 5210, PDI: 1.03. Tm = 36.5.
Example 58
Synthesis of ([G2]-PGLSA-MA)2-PEG - ([G2]-PGLSA-OH)2-PEG (0.501 g, 0.0863 mmol) was dissolved in DCM (15 mL) and stirred under nitrogen before methacrylic anhydride (0.50 mL, 3.36 mmol) was added by syringe. DMAP (72.1 mg, 0.990 mmol) was added and stirring was continued for 14 hours. Any remaining anhydride was quenched by the addition of methanol (0.1 mL, 3.95 mmol), which was allowed to stir for another 5 hours. The reaction was diluted with DCM (35 mL) and washed with 0.1 N HCl (50 mL) and brine (50 mL). The organic phase was dried with Na2SO4 and filtered before the PEG-based dendrimer was precipitated in cold (-20 0C) ethyl ether (300 mL) and collected to yield 0.534 g of a white solid (90 % yield). 1H NMR (CDCl3): δ 1.89 (m, 47, -
CH3), 2.60 (m, 65, -CH2-CH2-), 3.56-3.67 (broad m, 387, -CH2-CH2-), 3.77 (t, 2, -CH2- CH2-), 4.12-4.37 (broad m, 81, -CH2-CH-CH2-), 5.21 (m, 13, -CH2-CH-CH2-), 5.33 (m, 7, - CH2-CH-CH2-), 5.56 (m, 16, CH), 6.06 (m, 16, CH). 13C NMR (CDCl3): δ 171.89 (COOR), 135.84 (CH), 126.64 (CH), 70.75 (CH2), 69.45 (CH), 62.61 (CH2), 28.87 (CH2), 18.43 (CH3). FTIR: v (cm'1) 2873 (aliph. C-H stretch), 1736 (C=O). %). MALDI MS Mw: 6956, Mn: 6792, PDI: 1.02. SEC Mw: 4580, Mn: 4390, PDI: 1.04. Tm = 27.0.
Example 59
Synthesis of ([G3]-PGLSA-bzId)2-PEG - ([G2]-PGLSA-OH)2-PEG (2.13 g, 0.367 mmol), and 2-(m-l,3-0-benzylidene glycerol)succinic acid mono ester anhydride (12.71 g, 23.43 mmol) were dissolved in DCM (45 mL) and stirred under nitrogen. DMAP (0.608 g, 4.98 mmol) was added and stirring was continued for 14 hours. Any remaining anhydride was quenched by the addition of n-propanol (2.0 mL, 22 mmol), which was allowed to stir for another 5 hours. The reaction was diluted with DCM (55 mL) and washed with 0.1 N HCl (100 mL), saturated sodium bicarbonate (100 mL 3x), and brine (100 mL). The organic phase was dried with Na2SO4, filtered, and concentrated before the PEG-based dendrimer was precipitated in cold (-20 0C) ethyl ether (400 mL) overnight and collected to yield 3.35 g of a white solid (92 % yield). 1H NMR (CDCl3): δ 2.61 (broad m, 84, -CH2-CH2-), 2.74 (broad m, 36, -CH2-CH2-), 3.43 (t, 2, -CH2-CH2-), 3.56-3.65 (broad m, 278, -CH2-CH2-), 3.78 (t, 2, -CH2-CH2-), 4.13 (broad m, 60, -CH2-CH-CH2-), 4.21 (broad m, 60, -CH2-CH- CH2-), 4.69 (m, 16, -CH2-CH-CH2-), 5.19 (m, 14, -CH2-CH-CH2-), 5.50 (s, 16, CH), 7.32 (m, 46, arom. CH), 7.46 (m, 30, arom. CH). 13C NMR (CDCl3): δ 172.28 (COOR), 171.91 (COOR), 138.03 (CH), 129.26 (CH), 128.48 (CH), 126.21 (CH), 101.31 (CH), 70.76 (CH2), 69.49 (CH), 69.16 (CH2), 66.53 (CH), 62.47 (CH2), 29.35 (CH2), 29.02 (CH2), 28.83 (CH2). FTIR: v (cm"1) 2868 (aliph. C-H stretch), 1735 (C=O). MALDI MS Mw: 10215, Mn: 9985, PDI: 1.02. SEC Mw: 7020, Mn: 6900, PDI: 1.02. Tg = -13.6.
Example 60
Synthesis of ([G3]-PGLSA-OH)2-PEG - Pd(OH)2/C (10 % w/w) was added to a solution of ([G3]-PGLSA-bzld)2-PEG (2.88 g, 0.288 mmol) in 30 mL of 2:1 DCM/methanol. The apparatus for catalytic hydrogenolysis was evacuated and filled with 60 psi Of H2 before
shaking for 8 hours. The catalyst was filtered off and washed with DCM (20 mL). The PEG-based dendrimer was isolated after evaporation of solvents to give 2.86 g of a white solid (98 % yield). 1H NMR ((CD3)2CO):δ 2.64 (broad m, 120, -CH2-CH2-), 3.49-3.60 (broad m, 286, -CH2-CH2-), 3.64-3.75 (broad m, 33, -CH2-CH-CH2-), 4.00-4.12 (broad m, 42, -CH2-CH-CH2-), 4.13-4.29 (broad m, 68, -CH2-CH-CH2-), 4.64 (t, 2, -CH2-CH-CH2-), 4.85 (t, 2, -CH2-CH-CH2-), 5.26 (m, 14, -CH2-CH-CH2-). 13C NMR ((CD3)2CO): δ 171.85 (COOR), 171.64 (COOR), 76.09 (CH), 73.70 (CH2), 70.56 (CH), 69.52 (CH2), 66.19 (CH), 63.87 (CH2), 62.31 (CH2), 61.65 (CH2), 60.69 (CH2). FTIR: v (cm"1) 3432 (OH), 2925 (aliph. C-H stretch), 1734 (C=O). MALDI MS Mw: 8765, Mn: 8575, PDI: 1.02. SEC Mw: 8090, Mn: 7820, PDI: 1.03. Tg = -38.2.
Example 61
Synthesis of ([G3]-PGLSA-MA)2-PEG - ([G3] -PGLSA-OH)2-PEG (0.223 g, 0.0260 mmol) was dissolved in THF (15 mL) and stirred under nitrogen before methacrylic anhydride (1.10 mL, 7.38 mmol) was added by syringe. DMAP (90.0 mg, 0.737 mmol) was added and stirring was continued for 14 hours. Any remaining anhydride was quenched by the addition of methanol (0.2 mL, 7.89 mmol), which was allowed to stir for another 5 hours. The reaction was diluted with DCM (35 mL) and washed with 0.1 N HCl (50 mL) and brine (50 mL). The organic phase was dried with Na2SO4 and filtered before the PEG-based dendrimer was precipitated in cold (-20 0C) ethyl ether (300 mL) and collected to yield 0.248 g of a white solid (89 % yield). 1H NMR (CDCl3): δ 1.90 (m, 76, - CH3), 2.62 (m, 111, -CH2-CH2-), 3.56-3.67 (broad m, 285, -CH2-CH2-), 4.14-4.38 (broad m, 114, -CH2-CH-CH2-), 5.23 (m, 13, -CH2-CH-CH2-), 5.35 (m, 10, -CH2-CH-CH2-), 5.56 (m, 25, CH), 6.07 (m, 25, CH). 13C NMR (CDCl3): δ 171.87 (COOR), 135.91 (CH), 126.71 (CH), 70.76 (CH2), 69.47 (CH), 62.62 (CH2), 28.88 (CH2), 18.43 (CH3). FTIR: v (cm-1) 2874 (aliph. C-H stretch), 1734 (C=O). MALDI MS Mw: 10722, Mn: 10498, PDI: 1.02. SEC Mw: 7000, Mn: 6820, PDI: 1.03. Tg = -37.9.
Example 62
Synthesis of ([G4]-PGLSA-bzId)2-PEG - ([G3]-PGLSA-OH)2-PEG (1.82 g, 0.212 mmol), and 2-(cw-l,3-O-benzylidene glycerol)succinic acid mono ester anhydride (15.93 g,
29.36 mmol) were dissolved in THF (50 mL) and stirred under nitrogen. DMAP (0.537 g, 4.40 mmol) was added and stirring was continued for 14 hours. Any remaining anhydride was quenched by the addition of n-propanol (2.5 mL, 28 mmol), which was allowed to stir for another 5 hours. The reaction was diluted with DCM (50 mL) and washed with 0.1 N HCl (100 mL), saturated sodium bicarbonate (100 mL 3x), and brine (100 mL). The organic phase was dried with Na2SO4, filtered, and concentrated before the PEG-based dendrimer was precipitated in ethyl ether (400 mL) and collected to yield 3.11 g of a white solid (87 % yield). 1H NMR (CDCl3): δ 2.61 (broad m, 180, -CH2-CH2-), 2.70 (broad m, 64, -CH2-CH2-), 3.43 (t, 2, -CH2-CH2-), 3.56-3.65 (broad m, 286, -CH2-CH2-), 3.78 (t, 2, - CH2-CH2-), 4.11 (broad m, 125, -CH2-CH-CH2-), 4.23 (broad m, 125, -CH2-CH-CH2-), 4.68 (m, 32, -CH2-CH-CH2-), 5.20 (m, 30, -CH2-CH-CH2-), 5.49 (s, 32, CH), 7.32 (m, 93, arom. CH), 7.46 (m, 62, arom. CH). 13C NMR (CDCl3): δ 172.28 (COOR), 171.90 (COOR), 171.60 (COOR), 138.04 (CH), 129.26 (CH), 128.48 (CH), 126.21 (CH), 101.29 (CH), 70.76 (CH2), 69.46 (CH), 69.15 (CH2), 66.53 (CH), 62.57 (CH2), 29.34 (CH2), 29.18 (CH2), 29.02 (CH2), 28.83 (CH2). FTIR: v (cm"1) 2865 (aliph. C-H stretch), 1734 (C=O). MALDI MS Mw: 17289, Mn: 16968, PDI: 1.02. SEC Mw: 8110, Mn: 7950, PDI: 1.02. Tg = 5.3.
Example 63 Synthesis of ([G4] -PGLSA-OH)2-PEG - Pd(OH)2/C (10 % w/w) was added to a solution of ([G4]-PGLSA-bzld)2-PEG (2.88 g, 0.170 mmol) in 30 mL of 2:1 DCM/methanol. The apparatus for catalytic hydrogenolysis was evacuated and filled with 60 psi of H2 before shaking for 8 hours. The catalyst was filtered off and washed with DCM (20 mL). The PEG-based dendrimer was isolated after evaporation of solvents to give 2.86 g of a white solid (98 % yield). 1H NMR ((CD3)2CO):δ 2.64 (broad m, 248, -CH2-CH2-), 3.49-3.60 (broad m, 296, -CH2-CH2-), 3.66 (broad m, 50, -CH2-CH-CH2-), 3.82 (broad m, 42, -CH2- CH-CH2-), 4.04-4.16 (broad m, 66, -CH2-CH-CH2-), 4.28 (broad m, 124, -CH2-CH-CH2-), 4.86 (m, 10, -CH2-CH-CH2-), 5.27 (m, 30, -CH2-CH-CH2-). 13C NMR ((CD3)2CO): δ 172.20 (COOR), 70.45 (CH2), 70.10 (CH), 69.92 (CH2), 65.96 (CH), 62.31 (CH2). FTIR: v (cm"1) 3445 (OH), 2931 (aliph. C-H stretch), 1713 (C=O). MALDI MS Mw: 14402, Mn: 14146, PDI: 1.02. SEC Mw: 9130, Mn: 8980, PDI: 1.02. Tg = -18.0.
Example 64 Synthesis of bzld-[Gl]-PGLSA-TBDPS
4.00 g (0.014 mol) of bzld-[G I]-PGLSA-CO2H and 3.24 g (0.048 mol) of imidazole were stirred in 15 mL of DMF. Next, 6.4 niL (0.024 mol) of diphenyl-t-butyl silyl chloride were added and the reaction was stirred at 25 0C for 48 hours. The DMF was removed, the product was dissolved in CH2Cl2, washed with sat. NaHCO3 and water, dried over Na2SO4, filtered, rotovapped, and dried on the vacuum line. The product was purified by column chromatography (4:1 hexanes: EtOAc) affording 6.38 g of product as a viscous opaque oil (86% yield). Rf = 0.13 in 4:1 hexanes:EtOAc. 1H NMR (CDCl3): δ 1.09 (s, 9H, t-butyl), 2.78-2.84 (m, 4H, -CH2-CH2), 4.11-4.15 (m, 2Η, -CH2-CH-CH2-), 4.23-4.26 (m, 2Η, -CH2- CH-CH2-), 4.70-4.71 (m, 1Η, -CH2-CH-CH2-), 5.54 (s, IH, CH), 7.33-7.42, 7.48-7.50, 7.67-7.68 (m, 15Η, arom. bzld and phenyl CH) ppm. 13C NMR (CDCl3): δ 19.34 (-C- (CΗ3)3), 27.07 (-C-(CH3)3), 29.72, 30.96 (succ. -CH2-), 66.46, 69.18 (glycerol, 2C, -CH2-), 101.39 (0-CH-O), 126.23, 127.94, 128.50, 129.28, 130.29, 131.93, 135.51 (arom. CH), 137.99 (arom. bzld -C-), 171.53, 172.52 (succ. -C(=O)-) ppm. GC-MS: 519.2 m/z (MH+) (theory: 518.2 m/z (M+)). HR-FAB: 517.2028 m/z (M-H+) (theory: 518.2125 m/z (M+)). Elemental analysis: C, 69.18%; H, 6.69% (theory: C, 69.47%; H, 6.61%).
Example 65 Synthesis Of HO-[Gl]-PGLSA-TBDPS
2.41 g (4.65 mmol) of bzld-[G 1]-PGLSA-TBDPS was dissolved in 45 mL of THF, and 1.0 g of 20% Pd(OH)2/C was added. The solution was then placed in a Parr tube on a hydrogenator, evacuated, flushed with hydrogen, and shaken under 50 psi H2 for 3 hours. The solution was then filtered over wet celite. The product was purified by column chromatography (1:1 Hex:EtOAc increasing to 1:4 Hex:EtOAc) to yield 1.9 g of a clear oil (95% yield). 1H NMR (CDCl3): δ 1.08 (s, 9H, t-butyl), 2.02 (b s, 2H, -OH), 2.64-2.85 (m, 4Η, -CH2-CH2), 3.70-3.72, 4.07-4.14 (m, 4Η, -CH2-CH-CH2-), 4.83-4.86 (m, 1Η, -CH2- CH-CH2-), 7.33-7.44, 7.62-7.65 (m, 1OH, arom. phenyl CH) ppm. 13C NMR (CDCl3): δ 19.30 (-C-(CΗ3)3), 27.03 (-C-(CH3)3), 29.77, 31.37 (succ. -CH2-), 62.45 (glycerol, -CH2-), 75.86 (CH2-CH-CH2), 127.97, 130.36, 132.67, 135.49 (phenyl CH), 172.65, 178.24 (succ. - C(=O)-) ppm. FAB-MS: 431 m/z (M-H+) (theory: 430.57 m/z (M+)).
Acetyl derivative of compound HO-[G1]-PGLSA-TBDPS:
Compound HO-[G1]-PGLSA-TBDPS was a hydroscopic oil and repeated attempts to obtain satisfactory EA failed. Thus, we decided to prepare the acetyl analog for elemental analysis. 0.44 g (1.02 mmol) Of HO-[Gl]-PGLSA-TBDPS was stirred in 30 mL of CH2Cl2 with 0.30 g (1.02 mmol) of DPTS, 0.15 mL (2.66 mmol) of freshly distilled acetic acid, and 0.63 g (3.07 mmol) of DCC. The solution was stirred at RT for 18 hours. The DCU precipitate was filtered and the solution was evaporated. A solution of 1 : 1 ethyl acetate:hexanes was added and impurities precipitated. The solution was filtered, concentrated and further purified by column chromatography (3:1 hexanes:EtOAc), to afford 0.44 g of product (83% yield). Rf = 0.19 (4:1 hexanes: EtOAc) 1H NMR (CDCl3): δ 1.08 (s, 9H, t-butyl), 1.87-1.93 (m, 6H, -CH3), 2.50-2.71 (m, 4Η, -CH2-CH2), 3.96-4.19 (m, 4Η, -CH2-CH-CH2-), 5.06-5.18 (m, 1Η, -CH2-CH-CH2-), 7.22-7.33, 7.51-7.56 (m, 1OH, phenyl CH) ppm. 13C NMR (CDCl3): δ 19.10 (-C-(CΗ3)3), 20.61 (OC-CH3), 26.82 (-C- (CH3)3), 29.14, 30.62 (succ. -CH2-), 62.12, 69.28 (glycerol, -CH2-), 127.71, 130.09, 131.65, 135.27 (arom. CH), 170.52, 171.19, 171.58 ( -C(=O)-) ppm. FAB-MS: 515.4 m/z (MH+) (theory: 514.6 m/z (M+)). Elemental analysis: C, 62.76%; H, 6.69% (theory: C, 63.01%; H, 6.66%). SEC: Mw = 547, Mn = 528, PDI = 1.04.
Example 66 Synthesis of bzld-[G2]-PGLSA-TBDPS
1.90 g (4.41 mmol) Of HO-[Gl]-PGLSA-TBDPS was stirred in 100 mL Of CH2Cl2 with 1.30 g (1 equiv; 4.41 mmol) of DPTS, 2.72 g (9.70 mmol; 2.2 equiv) of 2(cis- 1,3-0- benzylidene glycerol)succinic acid monoester, and 2.00 g (9.70 mmol; 2.2 equiv) of DCC. The solution was stirred at RT for 18 hours. The DCU precipitate was filtered off and the solution was evaporated. A solution of 1 :1 ethyl acetate:hexanes was added and impurities precipitated. The solution was filtered, concentrated and further purified by column chromatography (1 : 1 hexanes: EtOAc) to afford 3.70 g of product (88% yield). Rf = 0.216 (1 : 1 hexanes:EtOAc). 1H NMR (CDCl3): δ 1.08 (s, 9H, t-butyl), 2.57-2.79 (m, 12H, -CH2- CH2), 4.08-4.14, 4.16-4.22 (m, 12Η, -CH2-CH-CH2-), 4.70-4.71 (m, 2Η, -CH2-CH-CH2-), 5.21 (m, IH, CH), 5.49-5.54 (m, 1Η, CH), 7.32-7.41, 7.47-7.49, 7.64-7.58 (m, 20Η, arom. bzld and phenyl CH) ppm. 13C NMR (CDCl3): δ 19.31 (-C-(CΗ3)3), 27.04 (-C-(CH3)3),
28.98, 29.33, 30.81 (succ. -CH2-), 62.48, 66.50, 69.16, 69.43 (glycerol, -CH2-), 101.33 (O- CH-O), 126.22, 127.95, 128.49, 129.26, 130.32, 131.92, 135.49 (arom. CH), 138.02 (arom. bzld -C-), 171.93, 172.28 (succ. -C(=O)-) ppm. GC-MS: 955.3 m/z (MH+) (theory: 954.4 m/z (M+)). Elemental analysis: C, 64.35%; H, 6.29% (theory: C, 64.14%; H, 6.12%). SEC: Mw = 940, Mn = 930, PDI = 1.01.
Example 67 Synthesis of bzld-[G2]-PGLSA-acid
1.00 g (1.04 mmol) of of bzld-[G2]-PGLSA-TBDPS was dissolved in 75 mL of THF. Next, 1.25 g (3.96 mmol) of tetrabutylammonium fluoride trihydrate was added to the solution and it was stirred at RT for 1 hour. After one hour the reaction was complete as indicated by TLC. The solution was diluted with 25 mL of H2O and acidified with IN
HCl to a pH of 3. The product was extracted into CH2Cl2, dried over Na2SO4, concentrated and dried on the vacuum line. The product was purified by column chromatography (0-5% MeOH in CH2Cl2; Rf = 0.24) for 0.65 g of product (87% yield). 1H NMR (CDCl3): δ 2.55-
2.77 (m, 12H, -CH2-CH2), 4.10-4.17, 4.24-4.31 (m, 12Η, -CH2-CH-CH2-), 4.74-4.75 (m,
2Η, -CH2-CH-CH2-), 5.28-5.31 (m, IH, CH), 5.52-5.54 (m, 2Η, CH), 7.33-7.38, 7.47-7.49
(m, 10Η, arom. bzld CH) ppm. 13C NMR (CDCl3): δ 28.72, 29.03, 29.38 (succ. -CH2-),
62.68, 66.56, 69.16 (glycerol, -CH2-), 101.44 (O-CH-0), 126.23, 128.50, 129.33 (arom. CH), 137.75 (arom. bzld -C-), 172.67, 175.16 (succ. -C(=O)-) ppm. GC-MS: 715.2 m/z
(M-Hl (theory: 716.2 m/z (M+)). Elemental analysis: C, 58.71%; H, 5.82% (theory: C,
58.66%; H, 5.63%). SEC: Mw = 810, Mn = 800, PDI = 1.01.
Example 68 Synthesis of HO-[G2]-PGLSA-TBDPS
1.55 g (1.62 mmol) of of bzld-[G2]-PGLSA-TBDPS was dissolved in 40 mL of THF and 1.0 g of 20% Pd(OH)2/C was added. The solution was then placed in a Parr tube on a hydrogenator and shaken under 50 psi H2 for 4 hours. The solution was then filtered over wet celite, rotoevaporated, and purified by column chromatography (0-25% acetone in EtOAc) to yield 1.12 g of product (95% yield). Rf = 0.25 (1 :3 acetone:EtOAc). 1H NMR (CDCl3): δ 1.07 (s, 9H, t-butyl), 2.25 (b s, 4H, -OH), 2.58-2.82 (m, 12Η, -CH2-CH2), 3.71-
3.74, 4.09-4.26 (m, 12H, -CH2-CH-CH2-), 4.87-4.99, 5.24-5.25 (m, 3Η, -CH2-CH-CH2-), 7.34-7.43, 7.63-7.48 (m, 1OH, phenyl CH) ppm. 13C NMR (CDCl3): δ 14.52 (-C-(CΗ3)3),
25.78 (-C-(CH3W, 26"> 29-30> 30-51> 30-81 (succ- -CH2-). 62.08, 63.44, 68.17, 70.23 (glycerol, -CH2-), 125.71, 127.96, 130.35, 135.45 (phenyl), 171.94, 172.40 (succ. -C(=O)-) ppm. GC-MS: 779.5 m/z (MH+) (theory: 778.3 m/z (M+)). SEC: Mw = 800, Mn = 792, PDI =1.01 Acetyl derivative of HO-[G2]-PGLSA-TBDPS:
Compound HO-[G2]-PGLSA-TBDPS was a hydroscopic oil and repeated attempts to obtain satisfactory EA failed. Thus, we decided to prepare the acetyl analog for elemental analysis. 0.55 g (0.70 mmol) of of HO-[G2]-PGLSA-TBDPS was stirred in 40 mL Of CH2Cl2 with 0.39 g (1.34 mmol) of DPTS, 0.19 mL (3.36 mmol) of freshly distilled acetic acid, and 0.87 g (4.20 mmol) of DCC. The solution was stirred at RT for 18 hours. The DCU precipitate was filtered and the solution was evaporated. The residue was resuspended in a minimum Of CH2Cl2, cooled to 10 0C and filtered. The resulting solution was concentrated and further purified by column chromatography (0-5% acetone in CH2Cl2) to afford 0.49g of product (66% yield). Rf = 0.17 (5% acetone in CH2Cl2) 1H NMR (CDCl3): δ 1.07 (s, 9H, t-butyl), 2.04 (s, 12H, -CH3), 2.55-2.83 (m, 12Η, -CH2-CH2), 4.09-4.32 (m, 12Η, -CH2-CH-CH2-), 5.20-5.29 (m, 3Η, -CH2-CH-CH2-), 7.32-7.44, 7.61- 7.67 (m, 1OH, phenyl CH) ppm. 13C NMR (CDCl3): δ 19.10 (-C-(CΗ3)3), 20.67 (OC-CH3), 26.82 (-C-(CH3)3), 28.60, 28.80, 29.10, 30.59 (succ. -CH2-), 62.11, 62.31, 69.39 (glycerol, -CH2-), 127.72, 130.09, 131.67, 135.27 (arom. CH), 170.50, 171.33, 171.61 ( -C(=O)-) ppm. FAB-MS: 947.9 m/z (MH+) (theory: 947.0 m/z (M+)). Elemental analysis: C, 57.15%; H, 6.26% (theory: C, 57.07%; H, 6.17%). SEC: Mw = 1075, Mn = 1041 , PDI = 1.03.
Example 69 Synthesis of bzld-[G3]-PGLSA-TBDPS
The bzld-[G3]-PGLSA-TBDPS dendron was synthesized by two methods, first by coupling of a bzld-[G2]-PGLSA-acid dendron to a HO-[G1]-PGLSA-TBDPS dendron convergently, and second by coupling compound to a HO-[G2]-PGLSA-TBDPS dendron (7) divergently.
Convergently: 1.05 g (1.47 mmol) of bzld-[G2]-PGLSA-acid was stirred in 75mL of CH2Cl2, and 0.29 g (0.67 mmol) of HO-[G1]-PGLSA-TBDPS, 0.20 g (0.67 mmol) DPTS, and 0.41 g (2.00 mmol) DCC were added. The solution was stirred at RT for 48 hours. The DCU precipitate was filtered off and the solution was evaporated. The product was purified by column chromatography (3:7 hexanes: EtOAc, Rf = 0.08) with a yield of 0.99 g (82% yield).
Divergently: 0.55 g (0.71 mmol) of a HO-[G2]-PGLSA-TBDPS was stirred in 50 mL Of CH2Cl2, and 0.42 g (1.41 mmol) of DPTS, 0.871 g (3.1 1 mmol) of 2(cis- 1,3-0- Benzylidene Glycerol)Succinic Acid Monoester, and 0.64 g (3.12 mmol) of DCC were added. The solution was stirred under nitrogen at RT for 18 hours. The DCU precipitate was filtered and the solution was evaporated. The product was purified by column chromatography (3:7 hexanes: EtOAc) to afford 0.71 g of product (54% yield). Rf = 0.08 (3:7 hexanes:EtOAc). 1H NMR (CDCl3): δ 1.08 (s, 9H, t-butyl), 2.54-2.92 (m, 28H, -CH2- CH2), 4.08-4.15, 4.22-4.27 (m, 28Η, -CH2-CH-CH2-), 4.71 (s, 4Η, -CH2-CH-CH2-), 5.21- 5.24 (m, 3H, CH), 5.52 (s, 4Η, CH), 7.31-7.42, 7.42-7.49, 7.65-7.67 (m, 30Η, arom. bzld and phenyl CH) ppm. 13C NMR (CDCl3): δ 19.31 (-C-(CΗ3)3), 27.04 (-C-(CH3)3), 29.35, 30.81 (succ. -CH2-), 62.49, 66.53, 69.16, 69.47 (glycerol, -CH2-), 101.33 (O-CH-O), 126.21, 127.94, 128.48, 129.26, 130.32, 135.47 (arom. CH), 138.02 (arom. bzld -C-), 171.90, 172.28 (succ. -C(O)-) ppm. GC-MS: 1825.6 m/z (M-H+) (theory: 1827.9 m/z (M+)). HR-FAB: 1825.6124 m/z (M-H+) (theory: 1826.6233 m/z (M+)). Elemental analysis: C, 60.66%; H, 5.85% (theory: C, 61.11%; H, 5.85%). SEC: Mw = 1830, Mn = 1810, PDI = LOl.
Example 70 Synthesis of bzld-[G3]-PGLSA-acid
2.00 g (1.09 mmol) of bzld-[G3]-PGLSA-TBDPS was dissolved in 125 mL of THF. Next, 1.3 g (4.1 mmol) of tetrabutylammonium fluoride trihydrate was added to the solution. The mixture was stirred at RT for 1 hour. After one hour the reaction was complete as indicated by TLC. The solution was diluted with 25 mL Of H2O and acidified with IN HCl to a pH of 3. The product was extracted into CH2Cl2, dried over Na2SO4, rotoevaporated and dried on the vacuum line. The product was purified by column
chromatography (0-5% MeOH in CH2Cl2) to afford 1.44 g of product (83% yield). Rf = 0.21 (5% MeOH in CH2Cl2). 1H NMR (CDCl3): δ 2.58-2.75 (m, 28H, -CH2-CH2), 4.11- 4.16, 4.19-4.27 (m, 28Η, -CH2-CH-CH2-), 4.71-4.72 (m, 4Η, -CH2-CH-CH2-), 5.21-5.28 (m, 3H, CH), 5.52-5.53 (m, 4Η, CH), 7.32-7.37, 7.46-7.49 (m, 20Η, arom. bzld CH) ppm. 13C NMR (CDCl3): δ 29.05, 29.36 (succ. -CH2-), 62.51, 66.58, 69.16 (glycerol, -CH2-), 101.36 (O-CH-0), 126.21, 128.49, 129.29 (arom. CH), 137.95 (arom. bzld -C-), 171.83, 173.01 (succ. -C(=O)-) ppm. GC-MS: 1587.5 m/z (M-H+) (theory: 1588.5 m/z (M+)). Elemental analysis: C, 58.02%; H, 5.60% (theory: C, 58.18%; H, 5.58%). SEC: Mw = 1650, Mn = 1620, PDI = 1.02.
Example 71 Synthesis of HO-[G3]-PGLSA-TBDPS
0.53 g (0.29 mmol) of bzld-[G3]-PGLSA-TBDPS was dissolved in 50 mL of THF in a Parr tube. 0.4 g of 20% Pd(OH)2/C was added and the flask was evacuated and filled with 50 psi of H2. The mixture was shaken for 8 hours, then filtered over wet celite. The filtrate was dried to produce a clear oil which was purified by column chromatography (0- 50% acetone in EtOAc) to afford 0.38 g of product (88% yield). Rf = 0.23 (1 : 1 acetone:EtOAc). 1H NMR (CDCl3): $1.3 (s, 9H, t-butyl), 2.52-2.86 (m, 28H, -CH2-CH2), 3.44_3.94 (mj 24, -CH2-CH-CH2- and -OH), 4.10-4.38, (m, 12Η, -CH2-CH-CH2-), 4.82- 4.92 (m, 4Η, CH), 5.18-5.30 (m, 3Η, CH), 7.28-7.43, 7.50-7.54, 7.60-7.66 (m, 10Η, phenyl CH) ppm. 13C NMR (CDCl3): δ 19.04 (-C-(CΗ3)3), 24.44 (-C-(CH3)3), 26.76, 27.12, 28.82, 28.97, 29.10, 30.57 (succ. -CH2-), 61.17, 62.33, 63.21, 69.30, 75.52 (glycerol, -CH2- ), 127.72, 130.11, 131.57, 134.36, 135.20 (arom. CH), 171.66, 171.72, 171.99, 172.27, 172.38, 172.46 (succ. -C(=O» ppm. MALDI-MS: 1475.56 m/z (MH+) (theory: 1475.5 m/z (M+)). SEC: Mw = 2101, Mn = 1994, PDI = 1.05.
Acetyl derivative of compound of HO-[G3]-PGLSA-TBDPS:
Compound HO-[G3]-PGLSA-TBDPS was a hydroscopic oil and repeated attempts to obtain satisfactory EA failed. Thus, we decided to prepare the acetyl analog for elemental analysis. 0.24 g (0.16 mmol) of HO-[G3]-PGLSA-TBDPS was stirred in 40 mL of CH2Cl2 with 0.19 g (0.65 mmol) of DPTS, 0.09 mL (1.55 mmol) of freshly distilled acetic acid, and 0.40 g (1.94 mmol) of DCC. The solution was stirred at RT for 18 hours.
The DCU precipitate was filtered and the solution was evaporated. The residue was resuspended in a minimum Of CH2Cl2, cooled to 10 0C and filtered. The resulting solution was concentrated and further purified by column chromatography (8:2 hexanes:EtOAc to 3:7 hexanes:EtOAc) to afford 0.18 g of product (63% yield). Rf = 0.15 (3:7 hexanes:EtOAc) 1H NMR (CDCl3): δ 1.10 (s, 9H, t-butyl), 1.99 (s, 24H, -CH3), 2.48-2.78 (m, 28Η, -CH2-CH2), 4.02-4.30 (m, 28Η, -CH2-CH-CH2-), 5.12-5.26 (m, 7Η, -CH2-CH- CH2-), 7.25-7.38, 7.55-7.61 (m, 1OH, phenyl CH) ppm. 13C NMR (CDCl3): δ 18.87 (-C- (CH-O3), 20.46 (OC-CH3), 26.61 (-C-(CH3)3), 26.95, 28.47, 28.55, 28.64, 28.90, 30.39 (succ. -CH2-), 61.90, 62.10, 69.02, 69.22 (glycerol, -CH2-), 127.52, 129.90, 131.48, 135.05 (arom. CH), 170.26, 171.14, 171.40, 171.46 ( -C(=O)-) ppm. FAB-MS: 1812.2 m/z (MH+) (theory: 1811.8 m/z (M+)). Elemental analysis: C, 53.95%; H, 6.12% (theory: C, 53.70%; H, 5.90%). SEC: Mw = 1943, Mn = 1882, PDI = 1.03.
Example 72 Synthesis of bzId-[G4]-PGLSA-TBDPS
The bzld-[G4] -PGLSA-TBDPS dendron was synthesized by two methods, first by coupling of bzld-[G2]-PGLSA-acid dendron to a HO-[G2]-PGLSA-TBDPS dendron convergently, and secondly by coupling the monoester 2(cis-l,3-O-Benzylidene Glycerol)Succinic Acid Monoester to a HO-[G3]-PGLSA-TBDPS dendron divergently. Convergently: 0.14 g (0.18 mmol) of HO-[G2]-PGLSA-TBDPS was dissolved in
30 mL Of CH2Cl2. Next, 0.05 g (0.18 mmol) of DPTS, 0.82 g (1.10 mmol) of bzld-[G2]- PGLSA-acid and 0.22 g (1.10 mmol) of DCC were added. The solution was stirred at RT under nitrogen for 72 hours. The DCU was filtered, the filtrate was concentrated to dryness and the residue was resuspended in a minimum of cold THF. The solution was filtered, concentrated and purified by column chromatography (1 :1 hexanes:EtOAc to 1 :4 hexanes:EtOAc, Rf = 0.14) to afford 0.48 g of product (75% yield).
Divergently: 0.38 g (0.26 mmol) of HO-[G3]-PGLSA-TBDPS was dissolved in 50 mL Of CH2Cl2. Next, 1.00 g (3.57 mmol) of 2(cis-l,3-O-Benzylidene Glycerol)Succinic Acid Monoester, 0.10 g (0.34 mmol) of DPTS, and 0.656 g (3.57 mmol) of DCC were added to the mixture. The solution was stirred for 48 hours under nitrogen at RT. The DCU precipitate was filtered, concentrated and purified by column chromatography (1 :1
hexanes:EtOAc to 1 :4 hexanes:EtOAc, Rf = 0.14) to afford 0.572 g of product (60% yield). 1H NMR (CDCl3): δ 1.07 (s, 9H, t-butyl), 2.55-2.77 (m, 6OH, -CH2-CH2), 4.07-4.15, 4.22- 4.25 (m, 60Η, -CH2-CH-CH2-), 4.70 (s, 8Η, -CH2-CH-CH2-), 5.19-5.21 (m, 7H, CH), 5.51 (s, 8Η, CH), 7.30-7.40, 7.46-7.48, 7.63-7.65 (m, 50Η, arom. bzld and phenyl CH) ppm. 13C NMR (CDCl3): δ 14.40 (-C-(CΗ3)3), 27.03 (-C-(OLj)3), 29.02, 29.35 (succ. -CH2-), 62.47, 66.53, 69.16, 69.49 (glycerol, -CH2-), 101.31 (0-CH-O), 126.21, 127.94, 128.48, 129.26, 135.47 (arom. CH), 138.03 (arom. bzld -C-), 171.50, 171.90, 172.27 (succ. -C(=O)-) ppm. MALDI-MS: 3574.54 m/z (MH+) (theory: 3573.54 m/z (M+)). Elemental analysis: C, 59.49%; H, 5.70% (theory: C, 59.19%; H, 5.74%). SEC: Mw = 3420, Mn = 3350, PDI = 1.02.
Example 73 Synthesis of [G3]-PGLSA-bzld Dendrimer 0.019 g (0.084 mmol) of [GO]-PGLSA-OH, 12 was dissolved in 50 mL Of CH2Cl2.
Next, 0.64 g (0.40 mmol) of compound bzld-[G3]-PGLSA-acid, 0.074 g (0.25 mmol) of DPTS, and 0.10 g of DCC (0.50 mmol) were added. The solution was stirred for 72 hours at RT under nitrogen. The DCU was filtered off and the filtrate was concentrated. The additional DCU was precipitated in cold THF and filtered. The product was purified by column chromatography (0-5% MeOH in CH2Cl2) to yield 0.40 g of product (73% yield). 1H NMR (CDCl3): δ 2.60-2.74 (m, 116H, -CH2-CH2), 4.08-4.17 (m, 60Η, -CH2-CH-CH2-), 4.22-4.26 (m, 60Η, -CH2-CH-CH2-), 4.70 (s, 16Η, -CH2-CH-CH2-), 5.20-5.23 (m, 14H, CH), 5.51 (s, 16Η, CH), 7.32-7.36, 7.46-7.48 (m, 80Η, arom. bzld CH) ppm. 13C NMR (CDCl3): δ 29.02, 29.35 (succ. -CH2-), 62.47, 66.54, 69.16 (glycerol, -CH2-), 101.31 (O- CH-O), 126.21, 128.48, 129.26 (arom. CH), 138.01 (arom. bzld -C-), 171.83, 172.29 (succ. -C(O)-) PPm. MALDI: 6553.4 m/z (MH+) (theory: 6552.2 m/z (M+). Elemental analysis: C, 58.50%; H, 5.48% (theory: C, 58.29%; H, 5.57%). SEC: Mw = 4740, Mn = 4590, PDI = LOl .
Example 74 Synthesis of [G3]-PGLSA-OH Dendrimer, 14
0.33 g (0.051 mmol) of [G3]-PGLSA-bzld was dissolved in 50 mL of a 9:1 solution of THF and MeOH in a Parr tube. Next, 0.50 g of 20% Pd(OH)2/C was added and the flask was evacuated and filled with 50 psi of H2. The mixture was shaken for 7 hours, then filtered over wet celite. The filtrate was dried to produce 0.25 g of a clear oil (0.049 mmol, 97% yield). 1H NMR (CD3OD): 5 2.64 (m, 116, -CH2-CH2-), 3.51 (m, 26, -CH2-CH- CH2-), 3.67 (m, 28, -CH2-CH-CH2-), 3.80 (m, 12, -CH2-CH-CH2-), 4.05 (m, 14, -CH2- CH-CH2-), 4.14 (m, 14, -CH2-CH-CH2-), 4.22 (m, 22, -CH2-CH-CH2-), 4.30 (m, 22, - CH2-CH-CH2-), 5.26 (m, 14, -CH2-CH-CH2) ppm. 13C NMR (CD3OD): δ. 28.61 (CH2), 62.41 (CH2), 62.87 (CH2), 65.67 (CH2), 67.64 (CH), 69.91 (CH), 172.86 (COOR) ppm. MALDI-MS: 5144.8 m/z (MH+) (theory: 5142.5 m/z (M+)). Elemental analysis: C, 48.07%; H, 5.84% (theory: C, 48.11%; H, 5.84%). SEC Mw: 5440; Mn: 5370; PDI: 1.01.
Example 75
Synthesis of [G3] -PGLSA-MA Dendrimer (50% derivatized)
0.22 g (0.041 mmol) of [G3]-PGLSA-OH was dissolved in 5 mL of DMF. Next, 0.20 g (1.66 mmol) of DMAP was then added followed by 0.10 mL (0.67 mmol, 0.5 eq. to the peripheral hydroxyl groups on [G3]-PGLSA-OH) of freshly distilled methacrylic anhydride. After 4.5 hours the reaction was complete as indicated by TLC. 0.03 mL (0.67 mmol) of MeOH was added to the reaction and allowed to stir for an additional 20 minutes. The solution was precipitated into 300 mL of cold ethyl ether. The ether was decanted off and the remaining oily reside was diluted with 20 mL of CH2Cl2. The organic phase was washed with 1 N HCl and brine. The organic phase was dried over Na2SO4, flitered, and concentrated to approximately 2 mL. This concentrated solution was precipitated in 300 mL of cold ethyl ether. The ether was decanted off and the resulting oily residue was dried under reduced pressure to yield 0.20 g of product (78% yield). 1H NMR (CDCl3): δ 1.90 (s, 42H, -CH3), 2.55-2.77 (m, 116Η, -CH2-CH2), 3.61-3.78 (m, 30Η, -CH2-CH-CH2-), 4.07- 4.30 (m, 120H, -CH2-CH-CH2-), 5.58-5.62 (m, 16Η, =CH), 6.03-6.16 (m, 16Η, =CH) ppm. 13C NMR (CDCl3): δ 18.24 (-CH3), 29.56, 29.75 (succ. -CH2-), 61.52, 62.09, 62.14, 65.17, 65.83, 69.39, 69.56, 70.04, 73.23, 75.89 (glycerol -CH2-), 171.04, 171.25, 171.37, 171.58,
171.79, 172.14, 172.51 ppm. MALDI-MS: 6224.6 m/z (MH+) (theory: 6231.6 m/z (M+)). SEC: Mw = 3525, Mn = 2708, PDI = 1.30.
Example 76 Synthesis of bzld-[G3]-PGLSA-PEG-OMe
0.29 g (0.18 mmol) of bzld-[G3]-PGLSA-acid was dissolved in 75 niL Of CH2Cl2. Next 0.45 g (0.09 mmol) of 5000 MW polyethylene glycol) mono-methyl ether (PEG- OMe; MALDI-MS: Mw = 5147, Mn = 5074, PDI = 1.01), 0.037 g (0.18 mmol) of DCC, and 0.026 g (0.09 mmol) of DPTS were added to the solution. The solution was stirred under nitrogen at RT for 168 hours. The DCU was filtered and the filtrate was concentrated to dryness. The resulting residue was resuspended in THF, cooled, and the DCU was filtered. The resulting solution was precipitated in ethyl ether. The solid was dissolved in THF, stirred with Amberlyst A-21 ion-exchange resin (Aldrich) (weakly basic resin) to eliminate the excess 9. The solution was filtered and the filtrate was dried over Na2SO4, dissolved in CH2Cl2, washed with 0.1 N HCl, and dried over Na2SO4 to yield 0.53 g of a solid white product (89% yield). 1H NMR (CDCl3): δ 2.60-2.73 (m, 28H, -CH2-CH2), 3.36 (s, MME CH3) 3.57-3.64 (m, 406Η, PEG CH2), 4.11-4.26 (m, 28Η, -CH2-CH-CH2-), 4.71 (m, 4Η, -CH2-CH-CH2-), 5.21-5.23 (m, 3H, CH), 5.52-5.54 (m, 4Η, CH), 7.32-7.37, 7.46- 7.49 (m, 20Η, arom. bzld CH) ppm. 13C NMR (CDCl3): δ 29.36, 29.90 (succ. -CH2-), 62.48, 66.53, 69.17 (glycerol, -CH2-), 70.77 (PEG, -CH2-), 101.33 (0-CH-O), 126.21, 128.48, 129.26 (arom. CH), 137.80 (arom. bzld -C-), 171.90 (succ. -C(=O)-) ppm. MALDI-MS: Mw = 6671, Mn = 6628 PDI = 1.01 (theoretical MW = 6588). SEC: Mw = 6990, Mn = 6670, PDI = 1.04.
Example 77
Synthesis of HO- [G3] -PGLSA-PEG-OMe
0.52 g of bzld-[G3]-PGLSA-PEG-OMe was dissolved in 40 mL of THF. Next, 0.10 g of 20% Pd(OH)2/C was added. The reaction vessel was evacuated and flushed with hydrogen. The solution was shaken for 3 hours under 50 psi H2 at RT. The Pd(OH)2ZC was removed by filtering over wet celite. The filtrate was dried and precipitated in ethyl ether to yield 0.40 g of an opaque hydroscopic solid (83% yield). 1H NMR (CDCl3): δ 2.60-2.70
(m, 28H, -CH2-CH2), 3.36 (s, MME CH3) 3.53-3.78 (b m, 422Η, PEG CH2 and -CH2-CH- CH2-), 4.17-4.27 (m, 12Η, -CH2-CH-CH2-), 4.92 (m, 4Η, -CH2-CH-CH2-), 5.21-5.23 (m, 3H, CH) ppm. 13C NMR (DMSO): δ 29.14, 29.36 (succ. -CH2-), 60.25 (-CH3 OMe), 63.22, 66.54, 69.87 (glycerol, -CH2-), 70.43 (PEG, -CH2-), 172.35, 172.57 (succ. -C(=O)-) ppm. MALDI-MS: Mw = 6302, Mn = 6260, PDI = 1.01 (theoretical MW = 6136). SEC: Mw = 6660, Mn = 6460, PDI = 1.03.
Example 78 Synthesis of MA- [G3] -PGLSA-PEG-OMe 0.39 g (0.064 mmol) of HO-[G3]-PGLSA-PEG-OMe was dissolved in 30 mL of
CH2Cl2. Next, 10 mg (0.08 mmol) of DMAP and 0.15 mL methacrylic anhydride (1.0 mmol) were added and the solution was stirred at RT under nitrogen overnight. The solution was then washed with 0.1 N HCl, dried over Na2SO4, condensed, and precipitated in ether to afford 0.41 g of product (96% yield). 1H NMR (CDCl3): δ 1.92 (s, 24 H, -CH3- methacrylate), 2.63 (m, 28H, -CH2-CH2), 3.36 (s, MME CH3) 3.59-3.67 (m, 406Η, PEG CH2), 4.19-4.39 (m, 28Η, -CH2-CH-CH2-), 5.24 (m, 4Η, -CH2-CH-CH2), 5.35 (m, 3H, CH), 5.59 (s, 8Η, -CH2- methacrylate), 6.10 (s, 8Η, -CH2- methacrylate) ppm. MALDI-MS: Mw = 7080, Mn = 7008, PDI = 1.01 (theoretical MW = 6780). SEC: Mw = 6918, Mn = 6465, PDI = 1.07.
Example 79 Synthesis of Myr-[G2]-PGLSA-TBDPS
0.45 g (0.58 mmol) of compound OΗ-[G2]-PGLSA-TBDPS was dissolved in 75 mL of CH2Cl2 with 0.63 g (2.77 mmol) of myristic acid(Myr), 0.34 g (1.16 mmol) of DPTS, and 0.72 g (3.47 mmol) of DCC. The reaction was stirred at RT for 16 hours. The DCU precipitate was filtered and the solution was evaporated. The residue was resuspended in 50 mL of ethanol, cooled to 0 0C for 6 hours and filtered. The precipitate was resuspended in 75 mL Of CH2Cl2, washed with 75 mL Of H2O, dried over Na2SO4, and the solvent evaporated to yield 0.84 g of product (89% yield). 1H NMR (CDCl3): δ 0.80- 0.89 (t, 12H, -CH3), 1.08 (s, 9Η, t-butyl), 1.14-1.34 (m, 8OH, myristic -CH2-), 1.50-1.64 (m, 8Η, Q=O)-CH2-CH2-CH2-), 2.22-2.33 (t, 8H, C(=O)-CH2-CH2-), 2.53-2.83 (m, 12H,
succinic -CH2-CH2), 4.08-4.34 (m, 12Η, -CH2-CH-CH2-), 5.18-5.30 (m, 3Η, -CH2-CH- CH2-), 7.32-7.44, 7.61-7.67 (m, 1OH, phenyl CH) ppm. 13C NMR (CDCl3): δ 14.25, 22.67, 24.81, 26.85, 28.81, 28.79, 29.12, 29.24, 29.36, 29.53, 29.64, 31.97, 34.05, 61.88, 62.34, 69.17, 127.66, 130.13, 135.28, 138.77, 171.34, 171.69, 173.32 ppm. FAB-MS: 1620.1 m/z (MH+) (theory: 1620.29 m/z (M+)). Elemental analysis: C, 68.84%; H, 9.69% (theory: C, 68.94%; H, 9.58%). SEC: Mw = 2168, Mn = 2135, PDI = 1.02.
Example 80 Synthesis of Myr-[G2]-PGLSA-acid 0.81 g (0.50 mmol) of Myr-[G2]-PGLSA-TBPDS was dissolved in 100 mL of THF. Next, 0.55 g (1.75 mmol) of tetrabutylammonium fluoride trihydrate was added to the solution. The mixture was stirred at RT for 1 hour. After one hour the reaction was complete as indicated by TLC. The solution was diluted with 25 mL of H2O and acidified with IN HCl to a pH of 3. The product was extracted into EtOAc, dried over Na2SO4, rotoevaporated and dried on the vacuum line. The product was purified by column chromatography (0-3% MeOH in CH2Cl2) to afford 0.60 g of product (87% yield). Rf = 0.23 (3% MeOH in CH2Cl2). 1H NMR (CDCl3): δ 0.82-0.88 (t, 12H, -CH3), 1.20-1.31 (m, 80Η, myristic -CH2-), 1.53-1.64 (m, 8Η, -C(O)-CH2-CH2-CH2-), 2.26-2.33 (t, 8H, -C(O)- CH2-CH2-), 2.60-2.68 (m, 12H, -CH2-CH2-), 4.11-4.34 (m, 12Η, -CH2-CH-CH2-), 5.19-5.35 (m, 3Η, -CH2-CH-CH2-) ppm. 13C NMR (CDCl3): δ 14.16, 22.78, 24.98, 28.56, 28.87, 29.07, 29.24, 29.47, 29.63, 29.87, 32.01, 34.04, 62.02, 62.64, 69.16, 69.93, 171.47, 171.68, 173.51 ppm. FAB-MS: 1382.9 m/z (M-H+) (theory: 1381.9 m/z (M+)). Elemental analysis: C, 66.72%; H, 9.91% (theory: C, 66.92%; H, 9.92%). SEC: Mw = 2074, Mn = 2040, PDI = 1.02.
Example 81 Synthesis of 2-benzyl-l,3-di(Myr-[G2]-PGLSA)2-glycerol
0.85 g (0.62 mmol) of compound Myr-[G2]-PGLSA-acid was dissolved in 75 mL
Of CH2Cl2 with 0.05 g (0.26 mmol) of 2-benzyl-glycerol, 0.08 g (0.26 mmol) of DPTS, and 0.16 g (0.77 mmol) of DCC. The reaction was stirred at RT for 16 hours. The DCU precipitate was filtered and the solution was evaporated. The residue was resuspended in
50 mL of ethanol, cooled to 0 0C for 6 hours and filtered. The precipitate was purified by column chromatography (20-50% EtOAc in hexanes) to yield 0.63 g of product (85% yield). Rf = 0.17 (30% EtOAc in hexanes). 1H NMR (CDCl3): δ 0.81-0.88 (t, 24H, -CH3), 1.17-1.34 (m, 160Η, myristic -CH2-), 1.52-1.63 (m, 16Η, C(=O)-CH2-CH2-CH2-), 2.24-2.32 (t, 16H, C(=O)-CH2-CH2-), 2.58-2.66 (m, 24H, succinic -CH2-CH2), 3.77-3.85 (m, 1Η, - CH2-CH-CH2-), 4.04-4.38 (m, 28H, -CH2-CH-CH2-), 4.59-4.65 (s, 2Η, benzyl -CH2-), 5.17-5.34 (m, 6Η, -CH2-CH-CH2-), 7.25-7.34 (m, 5H, aromatic CH) ppm. MALDI-MS: 2933.4 m/z (M+Na+) (theory: 2933.0 m/z (M+Na4)). Elemental analysis: C, 67.92%; Η, 9.79% (theory: C, 67.69%; Η, 9.77%). SEC: Mw = 4388, Mn = 4258, PDI = 1.03.
Example 82 Synthesis of l,3-di(Myr-[G2]-PGLSA)2-glycerol
0.47 g (0.16 mmol) of 2-benzyl-l,3-di(Myr-[G2]-PGLSA)2-glycerol was dissolved in 20 mL of TΗF and 0.5 g of 10% Pd/C was added. The solution was then placed in a Parr tube on a hydrogenator and shaken under 50 psi H2 for 10 hours. The solution was then filtered over wet celite, rotoevaporated, to yield the product.
Example 83 Synthesis of bz-SA-[G2]-PGLSA-TBDPS 0.77 g (0.99 mmol) of compound HO-[G2]-PGLSA-TBDPS was dissolved in 75 mL Of CH2Cl2 with 0.99 g (4.76 mmol) of benzylated succinic acid (bz-sa), 0.58 g (1.98 mmol) of DPTS, and 1.23 g (5.91 mmol) of DCC. The reaction was stirred at RT for 16 hours. The DCU precipitate was filtered and the solution was evaporated. The residue was resuspended in a minimum Of CH2Cl2, cooled to 10 0C for 1 hour and filtered. The solution was concentrated under reduced pressure and purified by column chromatorgraphy (30- 50% EtOAc in hexanes) to afford 1.21 g of product (79% yield). Rf = 0.18 (40% EtOAc in hexanes). 1H NMR (CDCl3): δ 1.08 (s, 9H, t-butyl), 2.55-2.81 (m, 28H, succinic -CH2- CH2), 4.06-4.37 (m, 12Η, -CH2-CH-CH2-), 5.11 (s, 8Η, benzyl -CH2-), 5.18-5.29 (m, 3Η, - CH2-CH-CH2-), 7.22-7.44, 7.61-7.67 (m, 3OH, aromatic CH) ppm. 13C NMR (CDCl3): δ 19.13, 26.81, 28.42, 28.64, 28.70, 28.91, 29.07, 30.56, 62.68, 66.72, 69.07, 73.69, 127.68, 128.23, 128.54, 130.06, 131.73, 135.21, 135.77, 171.64, 171.73, 171.90 ppm. FAB-MS:
1539.6 m/z (MH+) (theory: 1539.7 m/z (M+)). Elemental analysis: C, 63.35%; H, 6.02% (theory: C, 63.19%; H, 5.89%).
Example 84 Synthesis of bz-SA-[G2]-PGLSA-acid
1.12 g (0.73 mmol) of bz-SA-[G2]-PGLSA-TBDPS was dissolved in 100 mL of THF. Next, 0.89 g (2.76 mmol) of tetrabutylammonium fluoride trihydrate was added to the solution. The mixture was stirred at RT for 1 hour. After one hour the reaction was complete as indicated by TLC. The solution was diluted with 25 mL of H2O and acidified with IN HCl to a pH of 3. The product was extracted into EtOAc, dried over Na2SO4, rotoevaporated and dried on the vacuum line. The product was purified by column chromatography (0-3% MeOH in CH2Cl2) to afford 0.71 g of product (75% yield). Rf = 0.18 (3% MeOH in CH2Cl2). 1H NMR (CDCl3): δ 2.54-2.69 (m, 28H, -CH2-CH2), 4.11- 4.31 (m, 12Η, -CH2-CH-CH2-), 5.09 (s, 8Η, benzyl -CH2-), 5.18-5.25 (m, 3Η, -CH2-CH- CH2-), 7.25-7.36 (m, 2OH, aromatic CH) ppm. 13C NMR (CDCl3): δ 28.57, 28.78, 28.94, 62.28, 62.43, 66.60, 69.16, 69.37, 128.24, 128.29, 128.61, 128.57, 171.33, 171.79, 171.95 ppm. FAB-MS: 1301.5 m/z (M-H+) (theory: 1301.3 m/z (M+)). Elemental analysis: C, 60.23%; H, 5.81% (theory: C, 60.00%; H, 5.58%). SEC: Mw = 1415, Mn = 1379, PDI = 1.03.
Example 85 Synthesis of bz-SA-[G4]-PGLSA-TBDPS
0.07 g (0.08 mmol) of compound HO-[G2]-PGLSA-TBDPS was dissolved in 40 mL Of CH2Cl2 with 0.53 g (0.41 mmol) of bz-SA-[G2]-PGLSA-acid, 0.05 g (0.17 mmol) of DPTS, and 0.11 g (0.51 mmol) of DCC. The reaction was stirred at RT for 48 hours. The DCU precipitate was filtered and the solution was evaporated. The residue was resuspended in a minimum Of CH2Cl2, cooled to 10 0C for 1 hour and filtered. The solution was concentrated under reduced pressure and purified by column chromatorgraphy (30- 80% EtOAc in hexanes) to afford 0.40 g of product (80% yield). Rf = 0.18 (65% EtOAc in hexanes). 1H NMR (CDCl3): δ 1.07 (s, 9H, t-butyl), 2.53-2.81 (m, 124H, succinic -CH2- CH2), 4.10-4.31 (m, 60Η, -CH2-CH-CH2-), 5.09 (s, 32Η, benzyl -CH2-), 5.18-5.28 (m, 15Η,
-CH2-CH-CH2-), 7.25-7.41, 7.45-7.49, 7.61-7.66 (m, 9OH, aromatic CH) ppm. 13C NMR (CDCl3): δ 26.72, 28.52, 28.73, 28.87, 62.15, 66.43, 68.84, 69.16, 125.91, 127.64, 128.11, 128.33, 128.46, 130.01, 135.16, 135.66, 171.25, 171.54, 171.64, 171.81 ppm. MALDI-MS: XXX m/z (MH+) (theory: XXX m/z (M+)). Elemental analysis: C, 60.70%; H, 5.74% (theory: C, 60.34%; H, 5.63%). SEC: Mw = 5142, Mn = 5064, PDI = 1.02.
Example 86 Synthesis of bz-SA-[G4]-PGLSA-acid
0.22 g (0.04 mmol) of bz-SA-[G4]-PGLSA-TBDPS was dissolved in 12 mL of THF. Next, 0.04 g (0.13 mmol) of tetrabutylammonium fluoride trihydrate was added to the solution. The mixture was stirred at RT for 4 hours. The solution was diluted with 5 mL of H2O and acidified with IN HCl to a pH of 3. Additional THF was added dropwise to keep product in solution. The product was extracted into EtOAc, dried over Na2SO4, rotoevaporated and dried on the vacuum line. The product was purified by column chromatography (20- 100% EtO Ac in hexanes) to afford the product. 1H NMR (CDCl3): δ 2.46-2.84 (m, 124H, -CH2-CH2), 4.12-4.49 (m, 60Η, -CH2-CH-CH2-), 5.02-5.36 (m, 57Η, benzyl -CH2- and -CH2-CH-CH2-), 7.25-7.48 (m, 80H, aromatic CH) ppm. 13C NMR (CDCl3): δ 28.79, 28.93, 62.21, 66.51, 69.24, 127.64, 128.17, 128.52, 135.69, 171.34, 171.73, 171.91 ppm.
Example 87 Synthesis of ZLys(Z)-OPFP
DCC (5.45 g, 26 mmol) was added in five portions over 10 minutes to a solution of ZLys(Z)OΗ (10 g, 24 mmol) and 1.1 equiv of pentafluorophenol in freshly distilled CH2Cl2 (40 ml). The reaction mixture was stirred under N2 at 25 0C for 2 h, filtered to remove the insoluble urea, concentrated to ~ 20 ml under reduced pressure, and then stored at 4 0C for 2 h. An additional filtration removed further urea, and the filtrate was diluted with hexane (25 ml) and stored at 4 0C for 4h. The resultant white precipitate was collected by filtration, washed with DCM/hexane (1:2, 3x5 ml), and dried in vacuum; yield 13.37 g (98%).1H NMR (CDCl3): δ 1.46 (m, 2, CH2-CH2); 1.54 (m, 2, CH2-CH2); 1.84 (m, 1, CH2- CH); 2.00 (m, 1, CH2-CH); 3.19 (m, 2, CH2-NH); 4.67 (m, 1, CH2-CH); 4.8 (m, 1, NH);
5.03 (m, 2, CH2-O); 5.11 (s, 2, CH2-O); 5.54 (m, 1, NH); 7.3 (m, 10, arom CH). 19F NMR (CDC13): δ -162.26 (t, 2, CF); -157.60 (t, 1, CF); -152.72 (d, 2, CF). Elemental analysis: (theory: C, 57.93; Η, 4.34) found C, 58.12; Η, 4.40
Example 88
Synthesis of ZLys(Z)Lys(ZLys(Z))OMe
LysOMe. 2ΗC1 (1.43 g, 6 mmol) was dissolved in DMF (45 ml) with the DIEA (2.35 g, 18 mmol), and then the HOBT (2.25 g, 14 mmol) was added. After 5 minutes ZLys(Z)OPFP (12.5 g, 21mmol) in DCM (30 ml) was added at 0 0C for 10 minutes. The mixture was stirred for 24 h at RT under N2. After concentration under vacuum the mixture was dissolved in DCM (50 ml) washed with NaHCO3 (2x150 ml), water (2x150 ml) and then dried over NaSO4. The solvent was removed, and the mixture was precipitated in ether to lead a pure white compound 5.72 g (98%).Η NMR (CDCl3): δ 1.35-1.79 (m, 18, CH2- CH2); 2.87 (m, 1, CH2-NH); 3.13 (m, 4, CH2-NH); 3.40 (m, 1, CH2-NH); 3.63 (s, 3, CH3); 4.16 (m, 1, CH-NΗ); 4.34 (m, 1, CH-NΗ); 4.38 (m, 1, CH-NΗ); 4.88-5.02 (4 x s, 8, CH2- O); 5.13 (m, 1, CH2-NH); 5.28 (m, 1, CH2-NH); 5.94 (d, 1, CΗ-NH); 6.25 (d, 1, CΗ-NH); 6.88 (m, 1, CΗ2-NH);7.19-7.27 (m, 20, arom CH). 7.43 (d, 1, CΗ-NH). FAB MS: 953.4 m/z (MH+) (theory: 952.4 m/z (M+)). Elemental analysis: (theory: C, 64.27; H, 6.77; Ν, 8.82; O, 20.14) found C, 63.98; H, 6.79; Ν, 8.81; O, 20.39.
Example 89 Synthesis of LysLys(Lys)OMe» 4HCl
Pd/C (10% w/w) was added to a solution of ZLys(Z)Lys(ZLys(Z))OMe (1 g, lmmol) in MeOH (50ml). The flask for catalytic hydrogenolysis was evacuated and filled with 50 psi of H2 before shaking for 1O h. The catalyst was filtered and washed with MeOH (20 ml). The filtered was acidified with HCl gas. The acid solution was evaporated to give 578 mg of the white compound (98%).Η ΝMR (DMSO-d6): δ 1.36-1.81 (m, 18, CH2- CH2); 2.75 (m, 4, CH2-NH3 +); 3.12 (m, 2, CH2-NH); 3.65 (s, 3, CH3); 3.82 (m, 1, CH-NΗ); 3.98 (m, 1, CH-NΗ); 4.25 (m, 1, CH-NΗ); 8.20-8.45 (m, 12, NH3 +); 8.88 (t, 1, CH2-NH); 9.18 (d, 1, CΗ-NH). FAB MS: 417.4 m/z (MH+- 4HC1) (theory: 416.3 m/z (M+)). Elemental
analysis: (theory: C, 40.65; H, 7.72; Cl, 25.26; N, 14.97) found C, 40.31; H, 7.87; Cl, 25.10; N, 14.97.
Example 90 Synthesis of IsoCysOH
L-cysteine hydrochloride monohydrate (100 g, 0.569 mol) was refluxed in dry acetone (1.5 L) under dry nitrogen for 1.5 hours. The white precipitate was collected by filtration and refluxed a second time in dry acetone. Again the white solid was collected to yield 103.6 g of pure product (92 % yield).
Example 91 Synthesis of IsoCys(Boc)OH
To a suspension of IsoCysOH (144 g, 0.727 mol) and di-tert-butyl dicarbonate (206 g, 0.943 mol) in dry acetonitrile was added DIEA (140 mL, 0.803 mol). The suspension was allowed to stir for two days. Afterward, the acetonitrile was removed in vacuo, and the remaining oil was redissolved in ethyl ether and concentrated once more to an oily solid. The oily solid was again dissolved in ethyl ether and the amine salts were removed by vacuum filtration through Celite. The ethereal filtrate was washed with 0.1 N HCl (2x), water (2x), and brine (Ix), dried with sodium sulfate, and concentrated to a clear oilt which was dissolved in hexanes and concentrated to a white solid in vacuo. Crystalization from hexanes yielded 142 g of a white solid (75 % yield). FAB MS: 260.1 m/z (MH") (theory: 261.1 m/z (M+)). Elemental analysis: (theory:50.55; H, 7.33; N, 5.36; O, 24.49; S, 12.27) found C, 50.26; H, 7.30; N, 5.20; S, 12.11.
Example 92
Synthesis of IsoCys(Boc)OPFP
DCC (4.11 g, 20 mmol) was added in five portions over 10 min to a solution of IsoCys(Boc)OH (4.8 g, 18 mmol) and 1.1 equiv of pentafluorophenol (3.42, 20 mmol)in freshly distilled CH2Cl2 (25 ml). The reaction mixture was stirred under N2 at 25 0C for 2 h,
filtered to remove the insoluble urea, concentrated to ~ 20 ml under reduced pressure, and then stored at 4 °C for 2 h. An additional filtration removed further urea, and the product was crystallized from hot hexane. The resultant white precipitate was collected by filtration and dried in vacuum; yield 5.8 g (950Zo)1 1H NMR (CDCl3): δ 1.43 (s, 6, Boc CH3); 1.49 (s, 3, Boc CH3); 1.81 (s, 3, Isopr CH3); 1.87 (s, 3, Isopr CH3); 3.24 (d-d, 1, CH2); 3.43 (d-d, 1, CH2); 5.14 (d, 1, CH). 19F NMR (CDC13): δ -162.24 (t, 2, CF); -157.70 (t, 1, CF); -152.94 (d, 2, CF). GC MS: 445.0 m/z (M + NH4 +) (theory: 427.0 m/z (M+)). Elemental analysis: (theory: C, 47.77; H, 4.25; N, 3.28; S, 7.50) found C, 47.74; H, 4.19; N, 3.35; S, 7.48
Example 93
Synthesis of isoCys(Boc)Lys(isoCys(Boc))Lys(isoCys(Boc)Lys(isoCys(Boc)))OMe
LysLys(Lys)OMe (500 mg, 0.8 mmol) was dissolved in DMF (25 ml) with DIEA (550 mg, 4 mmol, and then HOBT (695 mg, 4 mmol) was added. After 5 minutes the IsoCys(Boc)OPFP, (2.78 g, 5.6 mmol) in DCM (21 ml) was added at 0 0C for 10 minutes. The mixture was stirred for 24 h at RT under N2. After concentration under vacuum the mixture was dissolved in DCM (40 ml) washed by NaHCO3 (2x100 ml), water (2x100 ml) and dried over NaSO4. Evaporation of organic solvent gave an oil that was purified by silica gel chromatography (DCM-MeOH = 96/4): yield 951 mg (74%).Η NMR (CDCl3): δ 1.19-1.68 (m, 18, CH2-CH2); 1.43 (s, 36, Boc CH3); 1.74 and 1.83 (2 x s, 24, Isopr CH3); 3.22 (m, 14, CH2-NH and CH2-S); 3.68 (s, 3, CH3-O); 4.29 (m, 1, CH-NΗ); 4.40 (m, 1, CH- NH); 4.49 (m, 1, CH-NH); 4.69 (m, 4, CH-N); 6.40-7.00 (m, 6, NH)-13C NMR (CDC13): δ 22.69-25.47 (CH2); 28.96-30.27 (CH3); 31.43 (CH2-S); 34.32; 37.11; 39.98; 52.74-53.32; 67.90; 72.00-74-10 (isopr Q; 82.05 (Boc Q; 152.32-154.23 (O-CO-NH); 163.17 (CO- OCH3); 171.58-173.07 (CO). FAB MS: 1389.6 m/z (MH+) (theory: 1388.6 m/z (M+)). HR MS: 1390.8784 m/z (MH+) (theory: 1390.8799 m/z (MH+)). Elemental analysis: (theory: C, 54.44; H, 7.83; N, 10.08; S, 9.23) found C, 53.93; H, 7.70; N, 9.92; S, 9.15.
Example 94
Synthesis of isoCysLys(isoCys)Lys(isoCysLys(isoCys))OMe»4TFA TFA (5 ml) was added in 10 portions over 10 min to a solution of isoCys(Boc)Lys(isoCys(Boc))Lys(isoCys(Boc)Lys(isoCys(Boc)))OMe, (600 mg, 0.4
mmol) in freshly distilled CH2Cl2 (30 ml) at O 0C. The reaction mixture was stirred under N2 for 25 0C for 2 h. The solvent was removed by vacuum, and the mixture was precipitated in ether to afford a pure white compound 417 mg (97 %).Η NMR (CD3OD): δ 1.39-1.81 (m, 18, CH2-CH2); 1.73 (s, 24, Isopr CH3); 3.13-3.31 (m, 10, CH2-NH); 3.56 (m, 4, CH2-S); 3.68 (s, 3, CH3-O); 4.27 (m, 1, CH-NΗ); 4.36 (m, 2, CH-NΗ); 4.56 (m, 4, CH- NH-C)-13C NMR (CD3OD): δ 24.83-24.96 (CH2); 29.43-30.71 (CH3); 32.87-33.74 (CH2- S); 36.71-36.95; 40.94-41.38; 53.68-56.18; 65.78; 75.55-75.78 (isopr C); 165.31 (CO- OCH3); 170.17-174.88 (CO). FAB MS: 990.4 m/z (MH+) (theory: 989.4 m/z (M+)). Elemental analysis: (theory: C, 42.38; H, 5.58; N, 9.69; S, 8.87) found C, 42.10; H, 5.77; N, 9.92; S, 9.01.
Example 95 Synthesis of CysLys(Cys)Lys(CysLys(Cys))OMe»4HCl isoCysLys(isoCys)Lys(isoCysLys(isoCys))OMe,(400 mg, 0.4 mmol) was dissolved in HCl IN-MeOH 50/50 (60 ml), and stirred under N2 at 25 0C for 4 h. The solvent was removed by vacuum, and the mixture was precipitated in ether to lead a pure white compound 350 mg (90 %).Η NMR (DMSO d-6): δ 1.30-1.76 (m, 18, CH2-CH2); 2.76-3.19
(m, 18, CH2-NH, CH2-SH and CH2-SH); 3.63 (s, 3, CH3-O); 4.02 (m, 2, CH-NH3 +); 4.13
(m, 2, CH-NH3 +); 4.18 (m, 2, CH-NH); 4.32 (m, 1, NH-CH-CO2CH3); 8.18, 8.47 and 8.81 (m, 18, NH and NH3 +). 13C NMR (DMSO d-6): δ 23.34-25.87 (CH2); 28.91; 31.99; 49.23;
52.49; 54.56; 167.36 (CO-OCH3); 172.26-178.39 (CO). FAB MS: 829.6 m/z (MH+)
(theory: 828.3 m/z (M+)). HR MS: 829.3581 m/z (MH+) (theory: 828.3478 m/z (M+)).
Elemental analysis: (theory: C, 38.19; H, 6.62; N, 14.37; S, 13.16) found: C, 37.99; H, 6.67;
N, 14.21.
Example 96 Synthesis of the (succinic acid)2-PEG
(OH)2-PEG (10 g, 3 mmol) was dissolved in pyridine (30 ml) with succinic anhydride (5.88 g, 60 mmol), and stirred under N2 at 25 0C for 4 h. The solvent was
removed by vacuum, and the mixture was precipitated in ether to afford a product 10.48 g (99 %).
Example 97 Synthesis of (succinic acid NHS)2-PEG i (succinic acid)2-PEG (Ig, 0.3mmol) was dissolved in DCM with EDCI and DMAP and N-hydroxysuccinimide was added. The reaction was stirred at RT for 24 hours and the product iosolated by precipitation. NMR obtained .
Example 98 Synthesis of (succinic acid cesium SaIt)2-PEG
(succinic acid)2-PEG (Ig, 0.3mmol) was dissolved in water and the pH was adjusted to 7.5 with CSCO3. The solvent was removed to obtain the pure compound (99%).
Example 99 Synthesis of (dimethyl acetal succinic ester)2-PEG
(dimethyl acetal succinic ester)2-PEG was prepared by reaction of (succinic acid cesium SaIt)2-PEG5(Ig, 0.3 mmol), with bromoacetaldehyde dimethyl acetal (133 μl, 1.2mmol) in DMF (5 ml) at 60 0C for 3 days. The solvent was removed by vacuum, and the mixture was precipitated in ether.
Example 100 Synthesis of (dialdehyde succinic ester)2-PEG
(dialdehyde succinic ester)2-PEG was obtain by treatment of (dimethyl acetal succinic ester)2-PEG, with TFA (5% H2O) in CH2Cl2 (1 :3) at room temperature for 20 minutes. The solvent was removed by vacuum, and the product was precipitated in ethyl ether.
Example 101 Synthesis of PEG-([G1]-PGLSA-NHS)2
PEG-([G1 J-PGLSA-OH)2 (1.03 g, 0.232 mmol), which was dried under vacuum at 80 0C for three hours, was then dissolved in CH2Cl2 (40 mL). EDCI, DMAP, and N- hydrosuccinimide were added and the reaction was stirred for 24 hours. The product was isolated by precipitatation in cold ethyl ether.
Example 102
Synthesis of PEG-(lys)2
(NH2)-PEG (1.0 g), which was dried under vacuum at 80 0C for three hours, was then dissolved in DMF (45 ml) with the DIEA (2.35 g, 18 mmol). HOBT was then added. After 5 minutes ZLys(Z)OPFP in DCM (30 ml) was added at 0 0C for 10 minutes. The mixture was stirred for 24 h at RT under N2. The reaction was then stopped and the product precipitated in ethyl ether. The Z groups were removed using Pd/C (10% w/w) and hydrogen gas. A solution of the intermediate was dissolved in MeOH (50 ml) and pour into the hydrogenation flask. The flask for catalytic hydrogenolysis was evacuated and filled with 50 psi of H2 before shaking for 1O h. The catalyst was filtered and washed with MeOH (20 ml). The product was isolated by precipitation in ethyl ether..
Example 103 Synthesis of PEG-(lys-succinate-NHS2)2
PEG-(lys)2 (1.0 g), which was dried under vacuum at 80 0C for three hours, was dissolved in CH2Cl2 (40 mL) and then succinic anahydride was added. The reaction was stirred for 24 hours and the succinic acid derivatized product was isolated by precipitation in ethyl ether. Next, this intermediate was dissolved in CH2Cl2 (40 mL) EDCI, DMAP, and N-hydrosuccinimide were added and the reaction was stirred for 24 hours. The product was isolated by precipitatation in cold ethyl ether.
Example 104
Preparation of a non-covalently crosslinked gel/network using (didodecane methyl amine)2-PEG
The (didodecane methyl amine)2-PEG was prepared in two steps by first treating (NH2)-PEG with 8 equivalents of bromododecane, 15 equivalents of NaCO3 in reflux
ethanol to obtain (didodecane amine)2-PEG. The (didodecane amine)2-PEG, 1, was then treated with methyl iodine to afford (didodecane methyl amine)2-PEG after precipitation in ether.
This cationic-hydrophobic linear polymer is likely to form a gel with the carboxylated terminated dendritic polymers.
Example 105
General Procedure for the Preparation of an Hydrogel Through Photocrosslinking ([GI]-PGLSA-MA)2-PEG Five microliters of solution containing 0.5% EY in HEPES buffer (0.1 M, 7.4 pH),
100 μL of 5.0 M TEA, and 1 μL of VP were mixed with 2 mL of a 55 % w/v solution of the dendritic polymer in HEPES buffer. Upon laser exposure (argon ion laser, λmax = 488 and 514 nm, 200 mW) for 60 s, the pink viscous liquid crosslinked into a clear, soft, flexible hydrogel. This reaction can be performed under a variety of concentrations of polymer to prepare gels with different physical and mechanical properties. The crosslinked process can be with a UV or visible light system.
Example 106
General Procedure for the Preparation of an Hydrogel Through Photocrosslinking [G3]-PGLSA-MA
Gels were prepared by dissolving [G3 J-PGLSA-MA, DMPA, and VP (1,000:10: 1 respectively) in CH2Ch. The polymer solution was exposed to UV light from a UVP BLAK-RAY long wave ultraviolet lamp for 5 minutes. This reaction can be performed under a variety of concentrations of polymer to prepare gels with different physical and mechanical properties. The polymer may be crosslinked with a UV or visible light absorbing system.
Example 107
General Procedure for the Preparation of an Hydrogel Through Photocrosslinking MA-[G3]-PGLSA-PEG-OMe
Five microliters of a solution of 0.5% EY in HEPES buffer (0.1 M, 7.4 pH), 100 μL of 5.0 M TEA, and 1 μL of VP were mixed with 2 mL of a 55 % w/v solution of the dendritic polymer in HEPES buffer. Upon laser exposure (argon ion laser, λmax = 488 and 514 nm, 200 mW) for 60 s, the pink viscous liquid crosslinked into a clear, soft, flexible hydrogel. This reaction can be performed under a variety of concentrations of polymer to prepare gels with different physical and mechanical properties. The polymer may be crosslinked with a UV or visible light absorbing system.
Example 108 General Procedure for the Preparation of a Hydrogel Through Treatment of LysLys(Lys)OMe with ([GI]-PGLSA-MA)2-PEG
The gel was prepared by mixing an aqueous solution of the LysLys(Lys)OMe dendron with the ([GI ]-PGLSA-MA)2-PEG. For example, the dendron dissolved at 33% w/w in phosphate buffer pH=8.2 (10 mg dendron in 20 μl) and the ([Gl]-PGLSA-MA)2- PEG was dissolved at 50% w/w (50 mg ([GI ]-PGLSA-MA)2-PEG in 50 μL) in the same buffer. These two solutions were mixed together to lead a gel. This reaction can be performed under a variety of concentarations of polymer to prepare gels with much different physical and mechanical properties.
Example 109 General Procedure for the Preparation of a Hydrogel Through Treatment of LysLys(Lys)OMe with PEG n-hydroxysuccinimide ((NHS)2-PEG)
The gel was prepared by mixing an aqueous solution of the LysLys(Lys)OMe dendron with the PEG NHS. For example, the dendron dissolved at 33% w/w in phosphate buffer pH=8.2 (10 mg dendron in 20 μl) and the PEG diNHS (commercially available, Mw = 3400) was dissolved at 55% w/w (50 mg PEG diNHS in 40 μL) in the same buffer. These two solutions were mixed together to lead a gel. Gelation was over in a few minutes. This reaction can be performed under a variety of concentarations of polymer to prepare gels with much different physical and mechanical properties.
Example 110
General Procedure for the Preparation of a Hydrogel Through Treatment of LysLys(Lys)OMe with PEG dimaleimide ((MAL)2-PEG)
The gel was prepared by mixing an aqueous solution of the LysLys(Lys)OMe dendron with the PEG-MAL. For example, the dendron dissolved at 33% w/w in phosphate buffer pH=8.2 (10 mg dendron in 20 μl) and the PEG dimaleimide (commercially available,
Mw = 3400) was dissolved at 55% w/w (50 mg PEG dimaleimide in 40 μL) in the same buffer. These two solutions were mixed together to lead a gel. Gelation occurs over 15 minutes. This reaction can be performed under a variety of concentarations of polymer to prepare gels with much different physical and mechanical properties.
Example 111
General Procedure for the Preparation of a Hydrogel Through Treatment of LysLys(Cys)Lys(CysLys(Cys))OMe»HCl with PEG diaidehdye ((CHO)2-PEG) or (dialdehyde succinic ester)2-PEG
The gel was prepared by mixing an aqueous solution of the
CysLys(Cys)Lys(CysLys(Cys))OMe»4HCl dendrons with the peg-dialdehyde or (dialdehyde succinic ester)2-PEG. For example, the dendron dissolved at 33% w/w in phosphate buffer pH=7 (10 mg dendron in 20 μl) and the PEG compound was dissolved at 55% w/w (50 mg PEG in 40 μl) in the same buffer. These two solutions were mixed together to lead a gel. Gelation occurs almost immediately. This reaction can be performed under a variety of concentarations of polymer to prepare gels with much different physical and mechanical properties.
Example 112
General Procedure for the Preparation of a Hydrogel Through Treatment of CysLys(Cys)Lys(CysLys(Cys))OMe«4HCl with PEG NHS ((NHS)2-PEG)
The gel was prepared by mixing an aqueous solution of the CysLys(Cys)Lys(CysLys(Cys))OMe«4HCl dendrons with the PEG-(NHS)2. For example, the dendron dissolved at 33% w/w in phosphate buffer pH=7 (10 mg dendron in 20 μl) and
the PEG compound was dissolved at 55% w/w (50 mg PEG in 40 μl) in the same buffer. These two solutions were mixed together to lead a gel. Gelation occurs almost immediately. This reaction can be performed under a variety of concentarations of polymer to prepare gels with different physical and mechanical properties.
Example 113
General Procedure for the Preparation of a Hydrogel Through the Formation of Disulfide Bonds
The gel was prepared by allowing a solution of 22 mg of CysLys(Cys)Lys(CysLys(Cys))OMe«4HCl in 40 μL in phosphate buffered solution to rest for one week. The solution forms a weak hydrogel.
Example 114
The mechanical properties of photocrosslinked gels composed of a [G3J-PGLSA dendrimer, a ([Gl]-PGLSA)2-PEG dendritic-linear hybrid macromolecule, a [G3]- PGLSA-PEG-OMe dendritic-linear hybrid macromolecule, a PEG (3400 Mw), and a PEG (8000 Mw) were determiend. These three biodendritic polymers were selected since they span a range of biodendrimer compositions and structures. Cylindrical constructs of the photocrosslinked hydrogels were prepared as previously described, and evaluated for their compressive and shear properties on a dynamic mechanical spectrometer. Results showed a minimal sensitivity of the shear properties to concentration for the ([G1]-PGLSA)2-PEG biodendrimer, with a much stronger sensitivity to the compressive moduli. The biodendrimer-based hydrogel showed highly elastic properties (δ= 7°) at 7.5% crosslinkable biodendrimer concentration and more visco-elastic behavior at 15 and 20% crosslinkable biodendrimer concentrations (S= 61°). The dynamic shear stiffness, however, varied little over the range of concentrations from 10-20%, with values less than 2 kPa. In contrast, the compressive modulus E showed a stronger concentration dependence ranging from 5 kPa at 7.5% to 21 kPa at 20% macromer concentration. There was a strong sensitivity of the shear stiffness to concentration, from G* < 1 kPa for the 20% crosslinkable ([G3]-PGLSA)-PEG biodendrimer concentration, to nearly 1000 kPa for the
50% concentration of the [G3]-PGLSA crosslinkable biodendrimer. All hydrogel networks formed from the [G3] biodendrimers were highly elastic. This illustrates a unique feature of the biodendrimer scaffolds, that the physical properties are a nonlinear function of the dendrimer "generation" and macromer concentration, reflecting an interaction between higher numbers of available crosslinking sites (i.e., higher generation), structure (dendrimer vs hybrid dendritic-linear polymer), size, and flexibility prior to crosslinking (e.g., ([G3]- PGLSA)-PEG vs ([Gl]-PGLSA)2-PEG vs [G3]-PGLSA)).
Table 2. Mechanical Properties of photocrosslinked gels.
% w/v Gel ϋeq |G*|
/kPa (KPa) δ / °
20 % ([GI]-PGLSA-MA)2-PEG 34 1.7 61
15 % ([GI]-PGLSA-MA)2-PEG 20 1.7 61
10 % ([GI]-PGLSA-MA)2-PEG 6.4 1.3 14
7.5 % ([GI]-PGLSA-MA)2-PEG 3.7 1.2 7
50% [G3]-PGLSA-MA 940 7
20 % ([G3]-PGLSA-MA)-PEG 0.21
* Data collected at 10 rad/sec.
Example 115 Primary chondrocytes from the femoral condyles of skeletally immature pigs (3 - 5 months) were isolated and re-suspended (1 x 106 cells per mL in PBS) with biodendrimer solutions (7.5 % or 15 % w/v; ([G1]-PGLSA-MA)2-PEG) containing the biocompatible eosin-sensitized initiator system. The mixture was subsequently crosslinked in a cylindrical mold with an argon-ion surgical laser (solution also contained ethyl eosin and triethanol amine (TEA) as photoinitiator and co-catalyst; 60s pulsed at 200 mW), resulting in complete cell encapsulation. The cell-hydrogel constructs were subsequently cultured for 2 and 4 weeks in chondrocyte growth medium (4 mL, Ham's F-12, 10 % FBS, 50 μg/mL ascorbic acid, 100 μg/mL streptomycin) at 37 0C in 5 % CO2.
At each time point individual constructs were processed for paraffin embedding, sectioned and stained with H&E, Safranin-O, and Masson's trichrome. The cells encapsulated in the hydrogel retained their rounded morphology and were heterogeneously distributed throughout the hydrogel at all time points for both macromer concentrations. For the 7.5% w/v crosslinkable biodendrimer, sections at 2 and 4 weeks stained positively for Safranin-O, indicating the presence of glycosaminoglycans (GAG) in the vicinity of the cells and eventually, intense staining throughout most regions of the hydrogel scaffold. These sections also stained strongly with Masson's Trichrome, showing the accumulation of newly synthesized collagenous proteins, first pericellularly and eventually throughout the scaffold. In similar sections at 15% w/v crosslinkable biodendrimer, only weak staining with Safranin-O and Trichrome was observed. Immunostaining of the 7.5% hydrogels for type II collagen (DSHB, II-II6B3) demonstrated that chondrocytes expressed high levels of type II collagen, similar to native cartilage. While these results provide support for the superior chondrogenic capacity of the 7.5% cell-hydrogel constructs as compared to the 15% constructs in vitro, there was evidence of significant degradation in the 7.5% hydrogels compared to the 15% w/v hydrogels. Importantly these results demonstrate that the dendrimer-based hydrogel is a scaffold for cartilage repair.
Example 116 General. All chemicals and culture media were used as received and were stored at room temperature, in the dark, or 4 0C where appropriate. All errors are reported as data ± one standard deviation. All macromer concentrations were reported as percentage weight macromer per weight solvent; macromer solutions showed densities of 1.04 ± 0.02, 1.03 ± 0.01, and 1.04 ± 0.03 for solutions at 7.5, 10, and 15 % macromer 1 in PBS, respectively. Chondrocyte growth medium consisted of DMEM supplemented with 10 % FBS, 2.5 mg/mL phosphate-C, and 5 mL penicillin/streptomycin (Invitrogen, Carlsbad CA). Washing medium consisted of DMEM high glucose with L-glutamine, 110 mg/mL sodium pyruvate with pyridoxine HCl, 3.3 mIVL 300x stock gentamycin, 10 mL/L 10Ox stock kanamycin, and 5 mL/L 20Ox fungizone (Invitrogen, Carlsbad CA).
Part I: Hydrogel Synthesis. The PEG340O-(PGLS A-MA4J2 macromer 1 (Figure 11) was synthesized from poly(ethylene glycol) with an weight average molecular weight of 3400 g/mol as a core and biodendrimers based on glycerol (GL) and succinic acid (SA), as described previously by Carnahan et al. See Carnahan, M. A.; Middleton, C; Kim, J.; Kim, T.; Grinstaff, M. W., J. Am. Chem. Soc. 2002, 124, 5291-5293. In order to evaluate the mechanical properties of the crosslinked biodendrimer macromer 1 solutions at 7.5, 10, 15, and 20 % w/w in PBS (Dulbecco's Phosphate Buffered Saline, Invitrogen) were mixed with 5 % photoinitiator solution (0.1 % eosin-Y, 4 % N-vinyl-2-pyrrolidinone, and 40 % triethanolamine in PBS, Invitrogen). The resulting solution was subsequently crosslinked in cylindrical molds (ø 8 mm, h = 2 mm) with long-wave UV (30 minutes), a filtered Xenon arc lamp (2 minutes with a Spectra-Physics/Oriel, 300W Xe lamp with filter #59070, yielding —100 mW at 510 nm), or an Argon laser (60 seconds at 514 nm, 200 mW, Ultima SE 120V, Lumenis), to form a three-dimensional turgid hydrogel. The resulting pellets were stored in PBS at room temperature or 37 0C. Due to the absence of significant swelling, all concentrations reported here are initial macromer concentrations, uncorrected for swelling after crosslinking.
Part II: Hydrogel Swelling and Degradation. Cylindrical hydrogel samples for swelling and degradation testing were prepared by crosslinking biodendrimer solutions at 7.5, 10, and 15 % w/w (n = 3) with the eosin-based initiator mentioned in the previous section in cylindrical molds (ø 8 mm, h = 2 mm) with a filtered Xenon arc lamp (2 minutes with a Spectra-Physics/Oriel 300W Xe lamp with filter #59070, yielding -100 mW at 510 nm). The crosslinked hydrogel pellets were subsequently stored at 37 0C in phosphate buffered saline (PBS, Invitrogen) or chondrocyte cell culture medium, both supplemented with 0.1 % NaN3 to prevent bacterial or fungal infection. The weight of the samples was measured over a period of 35 days, and was normalized by the weight of the samples immediately after crosslinking. The weight change over time used as an estimate of swelling and hydrogel degradation in cell culture medium or PBS, in the absence of chondrocytes. The results are presented in Figure 12.
Part III: Mechanical Testing. Cylindrical hydrogel samples for dynamic mechanical testing were prepared by crosslinking biodendrimer solutions at 7.5, 10, 15, and 20 % w/w with the eosin-based initiator mentioned above in cylindrical molds (ø 8 mm, h = 2 mm)
with long-wave UV (30 minutes). The samples were removed from the molds and were subsequently allowed to equilibrate in PBS solution for 3 days at room temperature. After equilibration, the samples were investigated in a strain-controlled rheometer (ARES, Rheometrics Scientific) at 0.1 to 100 Hz (maximum strain amplitude of 0.05) with a parallel-plate geometry immersed in a PBS bath. No significant frequency dependence was detected in the observed frequency range. The frequency independent dynamic shear modulus I G* I , storage modulus G', loss modulus G", and the loss angle δ are consequently shown as averaged from experiments at 5 to 10 rad/s, allowing estimation of the errors from the standard deviation. Compressive stress-relaxation experiments were performed on similar samples up to 20 % compressive strain in increments of 5 % to determine the equilibrium compressive modulus. The compressive moduli were determined by linear regression of the equilibrium stress versus strain data; errors were estimated from the standard deviation. The results are presented in Figures 13 and 14.
Part IV: Chondrocyte Encapsulation. Chondrocytes were isolated from the femoral condyles of skeletally immature porcine knees (3-5 months) using an enzymatic digestion protocol described previously. See Kuettner, K.; Pauli, B.; Gall, G.; Memoli, V.; Schenk,
R., J. Cell. Biol. 1982, 93 (3), 743-50. Cells were washed with washing medium and were subsequently re-suspended at 2x107 cells/mL in either a 7.5 % w/w or 15 % w/w biodendrimer solution with 5 % of the photoinitiator system. Samples (100 μL) of the cell suspensions were placed in cylindrical molds (ø 8 mm, h = 2 mm) and crosslinked with an argon-ion laser (514 run, 200 mW, 60 sec, Ultima SE 120V, Lumenis, Santa Clara, CA) to create cell-gel constructs. These constructs were placed in individual wells and cultured in chondrocyte culture medium in a humidified atmosphere at 37 0C with 5 % CO2. The culture medium was replaced every 3 days. Unseeded constructs incubated under the same conditions served as controls. Constructs containing cells were harvested at 2, 4, and 12 weeks (n = 3) and processed for histology. Control constructs without chondrocytes were harvested at 12 weeks (n = 2) and studied similarly.
Part V: Histology and Immunohistochemistry. Cell-gel constructs were placed in paraformaldehyde for 30 minutes, followed by dehydration in a graded series of ethanol prior to embedding in paraffin. Paraffin-embedded sections were stained with H&E,
Safranin-0, or Masson's Trichrome for histological evaluation. Sections were also
immunolabelled for the presence of types I (Sigma C2456) and II (DSHB II-II6B3) collagen, with visualization via a horseradish-conjugated secondary antibody. The results are presented in Figure 15.
Example 117
E-Beam Sterilization of Hydrogel Sealant Formed from CysLys(Cys)Lys(CysLys(Cys))OMe«4HCl and PEG(NHS)2
The sealant formulation was sterilized using E-beam. After E-beam sterilization, the dendron and PEG(NHS)2 solutions formed a hydrogel sealant with 30 seconds. Thus, E-beam sterilization is an acceptable sterilization method.
Example 118
Antimicrobial Properties of Hydrogel Formed from
CysLys(Cys)Lys(CysLys(Cys))OMe«4HCl and PEG(NHS)2 Containing 0.005 wt% Polyhexamethylene biguanide (PHMB)
The antimicrobial properties of a hydrogel containing polyhexamethylene biguanide (PHMB) were tested by incubating the crosslinked adhesive/sealant with the organism Bacillus Atrophaeus (ATCC 9372) at a concentration of 10,000 cfu. The test employed a non-sterile adhesive, a sterile adhesive, and a positive control (N=5/group). After 24 hours, only the positive control supports the bacteria. Thus, the hydrogel with PHMB adhesive acts as barrier to bacteria.
Example 119
General Procedure for the Preparation of a Hydrogel Through Treatment of LysLys(Lys)OMe with tri-block hydrophilic/hydrophobic PEG-PPG-PEG n- hydroxysuccinimide ((NHS)2-PEG-PPG-PEG)
A gel was prepared by mixing an aqueous solution of LysLys(Lys)OMe dendron with PEG-PPG-PEG(NHS)2 (Mn 2900; 40% by wt of PEG). Specifically, the dendron was dissolved in a phosphate buffer at pH=9, and the poly(ethylene glycol)-poly(propylene glycol)-poly(ethylene glycol)-diNHS (abreviated as PEG-PPG-PEG(NHS)2) was dissolved in the same buffer but with a pH=8 such that the total weight percent was 15%. These two
solutions were mixed together to afford a gel. Gelation was over in a few minutes. This reaction can be performed under a variety of concentrations of polymer to prepare gels with substantially different physical and mechanical properties.
Example 120
General Procedure for the Preparation of a Hydrogel Through Treatment of LysLys(Lys)OMe with tri-block hydrophilic/hydrophobic PEG-PPG-PEG n- hydroxysuccinimide ((NHS)2-PEG-PPG-PEG) and PEG(NHS)2
A gel was prepared by mixing an aqueous solution of LysLys(Lys)OMe dendron with PEG-PPG-PEG(NHS)2 (Mn 2900; 40% by wt of PEG) and PEG(NHS)2 (Mn 3400). Specifically, the dendron was dissolved in a phosphate buffer at pH=9, and PEG-PPG- PEG(NHS)2 and PEG(NHS)2 were dissolved in the same buffer, but with a pH=8 such that the total weight percent was 15%. These two solutions were mixed together to afford a gel. Gelation was over in a few minutes. This reaction can be performed under a variety of concentrations of polymer to prepare gels with substantially different physical and mechanical properties.
Example 121
General Procedure for the Preparation of a Hydrogel Through Treatment of CysLys(Cys)Lys(CysLys(Cys))OMe»4HCl with tri-block hydrophilic/hydrophobic PEG-PPG-PEG n-hydroxysuccinimide ((NHS)2-PEG-PPG-PEG)
A gel was prepared by mixing an aqueous solution of
CysLys(Cys)Lys(CysLys(Cys))OMe dendron with PEG-PPG-PEG(NHS)2 (Mn 2900; 40% by wt of PEG). Specifically, the dendron was dissolved in a phosphate buffer at pH=8.2, and PEG-PPG-PEG(NHS)2 was dissolved in the same buffer such that the total weight percent was 15%. These two solutions were mixed together to afford a gel. Gelation was over in a few minutes. This reaction can be performed under a variety of concentrations of polymer to prepare gels with substantially different physical and mechanical properties.
Example 122
General Procedure for the Preparation of a Hydrogel Through Treatment of CysLys(Cys)Lys(CysLys(Cys))OMe*4HCl with tri-block hydrophilic/hydrophobic PEG-PPG-PEG n-hydroxysuccinimide ((NHS)2-PEG-PPG-PEG) and PEG(NHS)2 A gel was prepared by mixing an aqueous solution of
CysLys(Cys)Lys(CysLys(Cys))OMe dendron with PEG-PPG-PEG(NHS)2 (Mn 2900; 40% by wt of PEG) and PEG(NHS)2 (Mn 3400). Specifically, the dendron was dissolved in a phosphate buffer at pH=8.2, and PEG-PPG-PEG-(NHS)2 and PEG(NHS)2 were dissolved in the same buffer such that the total weight percent was 15%. These two solutions were mixed together to afford a gel. Gelation was over in a few minutes. This reaction can be performed under a variety of concentrations of polymer to prepare gels with substantially different physical and mechanical properties.
Example 123 General Procedure for the Preparation of a Hydrogel Through Treatment of
CysLys(Cys)Lys(CysLys(Cys))OMe»4HCl with the NHS-activated acid of Sebacic Acid (Sebacic Sulfo-n-hydroxysuccinimide ((NHS-SO3)2-SA)
A gel was prepared by mixing an aqueous solution of
CysLys(Cys)Lys(CysLys(Cys))OMe dendron with Sebacic Sulfo-n-hydroxysuccinimide. Specifically, the dendron was dissolved in a phosphate buffer at pH=8.2 and SA diNHS- SO3 was dissolved in the same buffer such that the total weight percent was about 30%. These two solutions were mixed together to afford a gel. Gelation was over in a few minutes. This reaction can be performed under a variety of concentrations of polymer to prepare gels with substantially different physical and mechanical properties.
Example 124
General Procedure for the Controlled Polymerization of a Hydrogel Through Combining the Powders CysLys(Cys)Lys(CysLys(Cys))OMe*4HCl and PEG(NHS)2, Co-dissolving the Powders in a Solution Designed to Yield a Final pH Close to pH 6.0,
and Passing the Solution Through an Ion Exchange Resin to Rasie the pH to a Level Appropriate for the Two Components to Quickly Crosslink (~pH 7-7.2).
To compare polymerization times, a first gel was prepared by mixing the solids CysLys(Cys)Lys(CysLys(Cys))OMe»4HCl (5.1 mg) and PEG(NHS)2 (36.0 mg, Mn 3400) in a syringe. A solution was prepared containing approximately 100 mM sodium phosphate dibasic and 30 mM sodium carbonate. 230 μl of the solution was drawn into the syringe containing the solids, pushed and pulled between the syringe and the buffer container to mix, and expressed through a cannula. The hydrogel set in approximately 1 minute.
A second gel was prepared by mixing the solids CysLys(Cys)Lys(CysLys(Cys))OMe»4HCl (5.1 mg) and PEG(NHS)2 (36.0 mg, Mn 3400) in a syringe. A solution was prepared of containing approximately 100 mM sodium phosphate dibasic and 30 mM sodium carbonate. 230 μl of the solution was drawn into the syringe containing the solids, pushed and pulled between the syringe and the buffer container to mix, and expressed through a cannula containing a plug of the anion exchange resin MTO-Dowex M43. The hydrogel set in approximately 25 seconds. In this example, the Dowex anion exchange resin serves to remove acid from the initial solution, raising the pH of the overall solution, and, therefore, increasing the rate of polymerization upon expression of the solution.
Incorporation by Reference All of the U.S. patents and U.S. patent application publications cited herein are hereby incorporated by reference.
Equivalents
Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments of the invention described herein. Such equivalents are intended to be encompassed by the following claims.