CN103140535A - 低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 - Google Patents
低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 Download PDFInfo
- Publication number
- CN103140535A CN103140535A CN2011800476845A CN201180047684A CN103140535A CN 103140535 A CN103140535 A CN 103140535A CN 2011800476845 A CN2011800476845 A CN 2011800476845A CN 201180047684 A CN201180047684 A CN 201180047684A CN 103140535 A CN103140535 A CN 103140535A
- Authority
- CN
- China
- Prior art keywords
- general formula
- epoxy resin
- compound
- expression
- epoxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 C*[C@@](*)(CO*OC)*C(CC1)(CC(CC2)C1CC2(*)C1(C)*CCCC*C1)C1(C)CCCCCC1 Chemical compound C*[C@@](*)(CO*OC)*C(CC1)(CC(CC2)C1CC2(*)C1(C)*CCCC*C1)C1(C)CCCCCC1 0.000 description 3
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/22—Di-epoxy compounds
- C08G59/24—Di-epoxy compounds carbocyclic
- C08G59/245—Di-epoxy compounds carbocyclic aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/02—Polycondensates containing more than one epoxy group per molecule
- C08G59/04—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof
- C08G59/06—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof of polyhydric phenols
- C08G59/063—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof of polyhydric phenols with epihalohydrins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/3218—Carbocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Epoxy Resins (AREA)
- Epoxy Compounds (AREA)
Abstract
本发明的目的在于获得在液晶中的溶解性受到抑制而可防止液晶的污染的、可作为高品质的密封剂的低聚物成分来使用的环氧树脂和部分酯化环氧树脂。本发明提供一种通过某种特定的结构对环氧树脂的结构进行改性而得的新的环氧树脂、和使环氧树脂发生部分酯化而得的部分酯化环氧树脂、以及它们的制造方法。
Description
技术领域
本发明涉及低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物。
背景技术
在液晶显示元件的制造方法中,滴下工艺是一种可通过在真空下将液晶直接滴下到密封剂的闭环内,使其贴合,进行真空开放,从而制成面板的工艺。此滴下工艺具有降低液晶的用量、缩短液晶向面板的注入时间等众多优点,作为目前使用大型基板的液晶面板的制造方法而言,已成为主流。在包括滴下工艺的方法中,涂布密封剂/液晶,使其贴合,然后进行间隙形成、位置调整,主要通过紫外线固化进行密封剂的固化。
目前,密封剂的描画位置完全处于光所照射的位置,因此现状是密封不易产生未固化的部分,因而不存在污染液晶的问题,但是近年来随着窄边化的需要,密封位置处于更接近显示像素的趋势。因此,由于来自密封剂的污染,所以处于对显示像素部的电特性造成影响而容易引起显示不良的趋势。特别是在小型面板中,从显示部至密封剂的距离窄,因污染性而导致的显示不良的发生更加明显。由此,需要对液晶污染少的密封剂。
到目前为止,作为降低对液晶的污染性的方法,进行了各种研究。
提出了通过使用结晶性环氧树脂,从而降低在制作液晶面板时的热固化过程中的树脂向液晶中的溶出的方案(专利文献1)。
作为环氧树脂,在为双酚S型、醚型、硫醚型和芴型环氧树脂并且具有环氧烷单元的化合物中,提出了抑制了液晶的污染性的密封剂(专利文献2)。
另外,作为密封剂的原料,还研究了使用部分酯化环氧树脂的情况。
提出了通过使3官能的环氧树脂或4官能的环氧树脂发生部分丙烯酰化,从而降低未发生丙烯酰化的化合物的比例,降低热固化时的溶出的方案(专利文献3)。
提出了通过利用具有羧基的(甲基)丙烯酸衍生物使3官能的苯酚酚醛清漆型环氧树脂或4官能的苯酚酚醛清漆型环氧树脂发生部分改性,从而以环氧树脂和丙烯酸树脂的配合物来提高液体稳定性,改善液晶的取向特性的方案(专利文献4)。
现有技术文献
专利文献
专利文献1:日本特开2006-23583号公报
专利文献2:日本特许第4211942号公报
专利文献3:日本特开2008-3260号公报
专利文献4:日本特开2008-179796号公报
发明内容
发明所要解决的课题
但是,由于专利文献1中记载的环氧树脂是结晶性的,所以存在为了用作液态密封剂而需要与液态树脂混合的可能性、或因相溶性而发生析出的可能性。
专利文献3和4中记载的3官能的环氧树脂和4官能的环氧树脂为高粘度,或者在多数情况下为固体物,因而作为密封剂的使用受限。
另外,就密封剂而言,在UV照射量低的情况下,有关污染性而言,在目前的情况下不足。
本发明的目的在于获得在液晶中的溶解性受到抑制且可防止液晶的污染的、可作为高品质密封剂的低聚物成分来使用的环氧树脂以及部分酯化环氧树脂。
用于解决课题的手段
本发明人等为了提高液晶面板用滴下密封材料所要求的特性,着眼于作为密封材料的主要成分的环氧树脂对液晶的污染性,进行了深入研究,结果发现,通过用某种特定结构对作为主要成分的环氧树脂的结构进行改性,从而可降低低聚物本身相在液晶中的溶解性/溶出性,从而完成了本发明。
即,本发明涉及一种环氧树脂,所述环氧树脂由通式(1)、通式(2)或通式(3)表示,
通式(1)、通式(2)和通式(3)中,
X为-O-、碳原子数1~4的烷撑基、或碳原子数2~4的烷叉基,
Y为碳原子数1~4的烷撑基-碳原子数6~20的亚芳基-碳原子数1~4的烷撑基、碳原子数1~4的烷撑基-碳原子数6~20的亚芳基、或由-R7-(O-R7)n-表示的基团(式中,R7为碳原子数1~4的烷撑基,n为0或1~6的整数),
R1、R2、R3、R4、R5和R6相互独立地为氢、缩水甘油基或甲基缩水甘油基,
各个R21各自相互独立地为氢或甲基,
R1、R2、R3、R4、R5和R6中的至少2个为缩水甘油基或甲基缩水甘油基。
本发明涉及一种部分酯化环氧树脂,所述部分酯化环氧树脂由通式(4)、通式(5)或通式(6)表示,

通式(4)、通式(5)和通式(6)中,
X、Y和R21如上述定义所述,
R11、R12、R13、R14、R15和R16为氢、缩水甘油基、甲基缩水甘油基、或由-Z-R8表示的基团(式中,Z为2-羟基丙撑基或2-甲基-2-羟基丙撑基,R8为丙烯酰基或甲基丙烯酰基),
R11、R12、R13、R14、R15和R16中的至少2个为缩水甘油基、甲基缩水甘油基、或由-Z-R8表示的基团,
缩水甘油基和甲基缩水甘油基、与丙烯酰基和甲基丙烯酰基的比例,即,缩水甘油基+甲基缩水甘油基/与丙烯酰基+甲基丙烯酰基,为10∶90~90∶10。
本发明涉及一种环氧树脂的制造方法,所述环氧树脂的制造方法包括工序(1A)~(1B),
工序(1A):使分子中具有2个以上的环氧基的多官能环氧化合物在金属催化剂的存在下与分子中具有2个以上的羟基的多羟基化合物发生反应,得到多官能环氧化合物的环氧开环体的工序,和
工序(1B):使在工序(1A)中得到的多官能环氧化合物的环氧开环体的羟基发生环氧化的工序。
本发明涉及根据上述所述的环氧树脂的制造方法,其是用于制造上述由通式(1)~通式(3)表示的环氧树脂的方法,包括下述工序(2A)~(2B),
工序(2A):使由通式(7a)、通式(8a)或通式(9a)表示的环氧化合物在金属催化剂的存在下与由下述通式(10)表示的二羟基化合物发生反应,得到由通式(7b)、通式(8b)或通式(9b)表示的环氧开环体的工序,
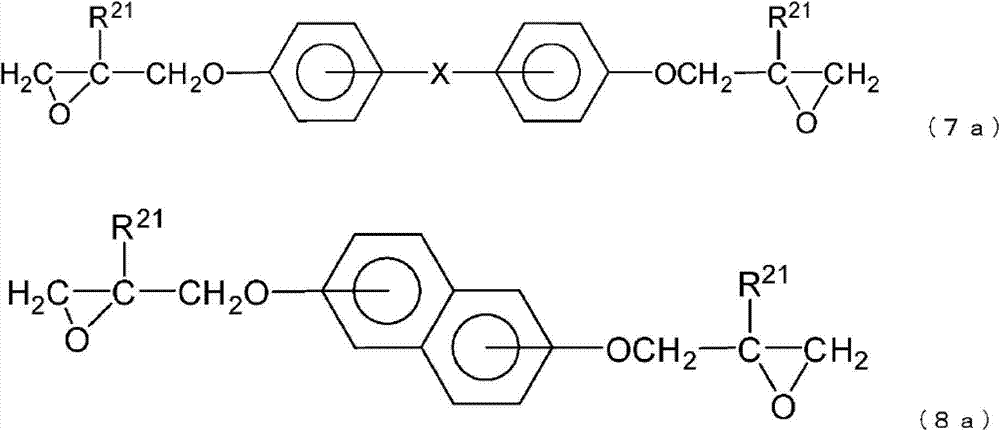

通式(7a)、通式(8a)和通式(9a)中,
X和R21如上述定义所述,
HO-Y-OH (10)
通式(10)中,Y如上述定义所述,

通式(7b)、通式(8b)和通式(9b)中,X、Y和R21如上述定义所述;和
工序(2B):使在工序(2A)中得到的由通式(7b)~通式(9b)表示的环氧开环体的羟基发生环氧化,得到由通式(1)~通式(3)表示的环氧树脂的工序。
本发明涉及一种部分酯化环氧树脂的制造方法,所述部分酯化环氧树脂的制造方法包括工序(1C),
工序(1C):使通过上述制造方法制得的环氧树脂在碱性催化剂的存在下与(甲基)丙烯酸发生反应的工序。
本发明涉及根据上述所述的部分酯化环氧树脂的制造方法,其是用于制造上述由通式(4)~通式(6)表示的部分酯化环氧树脂的方法,包括工序(2C),
工序(2C):使通过上述制造方法制得的由通式(1)、通式(2)或通式(3)表示的环氧树脂在碱性催化剂的存在下与(甲基)丙烯酸反应,得到由通式(4)~通式(6)表示的部分酯化环氧树脂的工序,


通式(1)、通式(2)和通式(3)中,
X、Y、R1、R2、R3、R4、R5、R6、R21如上述定义所述,
R1、R2、R3、R4、R5和R6中的至少2个为缩水甘油基或甲基缩水甘油基。
本发明涉及一种固化性组合物,所述固化性组合物含有选自(a)上述由通式(1)、通式(2)和通式(3)表示的环氧树脂,(b)上述由通式(4)、通式(5)和通式(6)表示的部分酯化环氧树脂,(c)通过上述制造方法制得的环氧树脂,以及(d)通过上述制造方法制得的部分酯化环氧树脂中的1种以上的树脂。
发明的效果
根据本发明,可获得在液晶中的溶解性受到抑制且可防止液晶污染的、可作为高品质密封剂的低聚物成分来使用的环氧树脂和部分酯化环氧树脂。
附图说明
图1是表示在照度1000mJ下的液晶的拐角部的取向性的显微镜图。
图2是表示在照度1000mJ下的液晶的直线部的取向性的显微镜图。
图3是表示在照度50mJ下的液晶的拐角部的取向性的显微镜图。
图4是表示在照度50mJ下的液晶的直线部的取向性的显微镜图。
图5是表示在照度0mJ下的液晶的拐角部的取向性的显微镜图。
图6是表示在照度0mJ下的液晶的直线部的取向性的显微镜图。
具体实施方式
以下对本发明的适宜的实施方式进行说明。
本发明的环氧树脂是由通式(1)、通式(2)和通式(3)表示的化合物。
碳原子数1~4的烷撑基可列举出亚甲基、乙撑基、丙撑基和丁撑基,优选为亚甲基和乙撑基。
碳原子数2~4的烷叉基可列举出乙叉基、丙叉基、异丙叉基、甲基丙叉基和丁叉基,优选为乙叉基和异丙叉基。
碳原子数6~20的亚芳基是单环或多环的芳基,可列举出亚苯基、亚萘基和亚蒽基,优选为亚苯基。
碳原子数1~4的烷撑基-碳原子数6~20的亚芳基中的碳原子数1~4的烷撑基和碳原子数6~20的亚芳基的例示如上述定义所述。作为碳原子数1~4的烷撑基-碳原子数6~20的亚芳基,可优选列举出亚甲基-亚苯基。在本发明中,与碳原子数1~4的烷撑基-碳原子数6~20的亚芳基中的各个基团的键合顺序可任选。例如,对于键合于R1~R6的氧原子而言,可以与碳原子数1~4的烷撑基-碳原子数6~20的亚芳基中的碳原子数1~4的烷撑基键合,还可以与碳原子数6~20的亚芳基键合。
碳原子数1~4的烷撑基-碳原子数6~20的亚芳基-碳原子数1~4的烷撑基中的碳原子数1~4的烷撑基和碳原子数6~20的亚芳基的例示如上述定义所述。作为碳原子数1~4的烷撑基-碳原子数6~20的亚芳基-碳原子数1~4的烷撑基,可优选列举出亚苯基双(亚甲基)。
在本发明中,R1~R6中的至少2个为缩水甘油基或甲基缩水甘油基。在本发明中,所谓环氧基包含缩水甘油基和甲基缩水甘油基这两者。在这里,缩水甘油基是2,3-环氧丙基,甲基缩水甘油基是2,3-环氧基-2-甲基丙基。在本发明中,优选R1~R6之中3个以上是缩水甘油基或甲基缩水甘油基,更优选R1~R6之中全部是缩水甘油基或甲基缩水甘油基。另外,在本发明中,优选缩水甘油基和甲基缩水甘油基存在于与R4、R5和R6即伯碳原子键合的氧原子上,更优选缩水甘油基和甲基缩水甘油基存在于与R1~R6即伯碳原子和仲碳原子键合的氧原子上。
在本发明中,由通式(1)~通式(3)表示的环氧树脂中的缩水甘油基和甲基缩水甘油基的数量可通过高效液相色谱法(HPLC)计算。具体而言,可通过HPLC得到与缩水甘油基和甲基缩水甘油基的个数相对应的峰,根据各自的峰面积算出缩水甘油基和甲基缩水甘油基的个数的存在比例。由此可算出化合物中含有的缩水甘油基和甲基缩水甘油基的个数。另外,在由通式(1)~(3)表示的环氧树脂是混合物的情况下,缩水甘油基和甲基缩水甘油基的数量可以以混合物的平均值的形式而算出。即,就环氧基的数量而言,可通过进行HPLC的各个峰的质量分析(LC-MS)来判断分子量,根据混合物中的各成分的存在比而算出混合物中的环氧基的平均数量。例如,存在由通式(1)和通式(2)表示的环氧树脂的混合物中包含环氧基为3个和4个的化合物及其多聚体,由通式(3)表示的环氧树脂中包含环氧基为3个、4个、5个和6个的化合物及其多聚体的情况。
由通式(1)表示的化合物的X的键合位置优选为4,4’-位,换言之,是具有双酚型环氧树脂骨架的化合物。另外,由通式(2)表示的化合物中的萘环的键合位置优选为1,6-键合。
在本发明中,由通式(1)~通式(3)表示的化合物的数均分子量优选为200~5,000。若在这样的范围,则粘接性良好,并且对液晶的污染性进一步得到抑制。在本发明中,数均分子量是采用凝胶渗透色谱法(GPC)通过聚苯乙烯换算而算出的数均分子量。
在本发明中,由通式(1)~通式(3)表示的环氧化合物的环氧当量优选为100~3,000g/eq.,更优选为200~1,000g/eq.。若在这样的范围,则粘接性良好,并且对液晶的污染性进一步得到抑制。在本发明中,在环氧树脂是混合物的情况下,环氧当量可以以混合物的平均值的形式而求出。在本发明中,环氧当量可根据JIS K 7236:2001(对应ISO3001:1999)来求出。
在本发明中,部分酯化环氧树脂由通式(4)、通式(5)和通式(6)表示。在由通式(4)~通式(6)表示的部分酯化环氧树脂中,R11、R12、R13、R14、R15和R16中的至少2个为缩水甘油基、甲基缩水甘油基、或含有(甲基)丙烯酰基的由-Z-R8表示的基团。即,本发明的由通式(4)~通式(6)表示的部分酯化环氧树脂是由通式(1)~通式(3)表示的环氧树脂的缩水甘油基和甲基缩水甘油基的一部分被(甲基)丙烯酰基化而得的环氧树脂。
在本发明中,优选R11~R16之中3个以上是缩水甘油基、甲基缩水甘油基、或含有(甲基)丙烯酰基的由-Z-R8表示的基团,更优选R11R16之中均为缩水甘油基、甲基缩水甘油基、或含有(甲基)丙烯酰基的由-Z-R8表示的基团。另外,在本发明中,优选缩水甘油基、甲基缩水甘油基和(甲基)丙烯酰基存在于与R14、R15和R16即伯碳原子键合的氧原子上,更优选缩水甘油基、甲基缩水甘油基和(甲基)丙烯酰基存在于与R11~R16即伯碳原子和仲碳原子键合的氧原子上。
由通式(4)~通式(6)表示的部分酯化环氧树脂中的缩水甘油基和甲基缩水甘油基的数量以及(甲基)丙烯酰基的数量可通过HPLC计算。具体而言,可通过HPLC得到与各个环氧基的数量和各个(甲基)丙烯酰基的数量相对应的峰,根据各自的峰面积算出各自个数的存在比例。由此求出由通式(4)~通式(6)表示的部分酯化环氧树脂中的缩水甘油基、甲基缩水甘油基、含有(甲基)丙烯酰基的由-Z-R8表示的基团的个数。另外,在由通式(4)~(6)表示的部分酯化环氧树脂是混合物的情况下,缩水甘油基和甲基缩水甘油基以及(甲基)丙烯酰基的数量可以以混合物的平均值的形式算出。例如,存在由通式(4)和通式(5)表示的部分酯化环氧树脂的混合物中包含由通式(1)和通式(2)表示的环氧树脂的混合物、以及它们的环氧基的一部分变为(甲基)丙烯酰基的树脂,由通式(6)表示的部分酯化环氧树脂包含由通式(3)表示的环氧树脂的混合物、以及它们的环氧基的一部分变为(甲基)丙烯酰基的树脂的情况。
在由通式(4)~通式(6)表示的部分酯化环氧树脂中,缩水甘油基和甲基缩水甘油基与丙烯酰基和甲基丙烯酰基的比例,即,环氧基与(甲基)丙烯酰基的比例,为10∶90~90∶10。在这里,环氧基与(甲基)丙烯酰基的比例可通过HPLC和环氧当量求出。具体而言,由于作为原料的环氧树脂的环氧当量只减少被部分酯化了的部分,所以可通过测定部分酯化环氧树脂的环氧当量,从而算出被酯化成何种程度。另外,可通过进行HPLC的各个峰的质量分析(LC-MS),而求出各种成分的分子量和存在比例,从而求出各种成分的环氧基和丙烯酰基的比例。
在本发明中,由通式(4)~通式(6)表示的部分酯化环氧树脂的数均分子量优选为500~10,000,更优选为800~5,000。另外,由通式(4)~通式(6)表示的部分酯化环氧树脂在20℃的粘度优选为1,000~1,000,000mP·s,更优选为40,000~600,000mP·s。若在这样的范围,则在涂布于液晶时不易发生流动,可抑制对液晶的污染。需说明的是,在本发明中,粘度是使用E型粘度计,在圆锥形转子的转速为2.5rpm下测得的值。
环氧树脂的制造方法
对本发明的环氧树脂的制造方法进行说明。在本发明的环氧树脂的制造方法中,包括以下工序:基于作为原料的多官能环氧化合物与多羟基化合物的反应而进行的多官能环氧化合物的环氧基的开环工序,和通过环氧基的开环所生成的环氧开环体的羟基、和源自多羟基化合物的羟基的环氧化工序。在本发明中,环氧化包含缩水甘油基化和甲基缩水甘油基化这两者。
本发明的环氧树脂的制造方法包括下列工序(1A)~(1B):
(1A)使分子中具有2个以上的环氧基的多官能环氧化合物在金属催化剂的存在下与分子中具有2个以上的羟基的多羟基化合物发生反应,得到多官能环氧化合物的环氧开环体的工序,和
(1B)使在工序(1A)中得到的多官能环氧化合物的环氧开环体的羟基发生环氧化的工序。
工序(1A)
在工序(1A)中,作为原料化合物的分子中具有2个以上的环氧基的多官能环氧化合物通过环氧基的开环而形成羟基,并且形成源自多羟基化合物的羟基。在这里,所谓多官能环氧化合物的开环体是指多官能环氧化合物的环氧基全部开环而得的化合物。
多官能环氧化合物只要是1分子中具有2个以上的环氧基的环氧化合物,就无特殊限定。作为多官能环氧化合物,可列举出以下化合物。
作为多官能环氧化合物,可列举出使乙二醇、二甘醇、三甘醇、丙二醇、一缩二丙二醇、二缩三丙二醇等多烷撑二醇类,二羟甲基丙烷、三羟甲基丙烷、螺环二醇、甘油等多元醇类与表氯醇发生反应而得的脂肪族多元缩水甘油醚化合物。
作为多官能环氧化合物,可列举出使双酚A、双酚S、双酚F、双酚AD等芳香族二醇类以及对它们进行乙二醇、丙二醇、烷撑二醇改性而得的二醇类与表氯醇发生反应而得的芳香族多元缩水甘油醚化合物。
作为多官能环氧化合物,可列举出使己二酸、衣康酸等芳香族二羧酸与表氯醇发生反应而得的脂肪族多元缩水甘油醚化合物,使间苯二甲酸、对苯二甲酸、均苯四酸等芳香族二羧酸与表氯醇发生反应而得的芳香族多元缩水甘油酯化合物。
作为多官能环氧化合物,可列举出使二氨基二苯基甲烷、苯胺、间苯二甲胺等芳香族胺与表氯醇发生反应而得的芳香族多元缩水甘油胺化合物。
作为多官能环氧化合物,可列举出使乙内酰脲及其衍生物与表氯醇发生反应而得的乙内酰脲型多元缩水甘油基化合物。
作为多官能环氧化合物,可列举出使由苯酚或甲酚与甲醛衍生出的酚树脂、酚醛清漆树脂与表氯醇发生反应而得的酚型多元缩水甘油醚化合物、酚醛清漆型多元缩水甘油醚化合物。
在本发明中,作为多官能环氧化合物,优选由下列通式(7a)、通式(8a)或通式(9a)表示的环氧化合物,

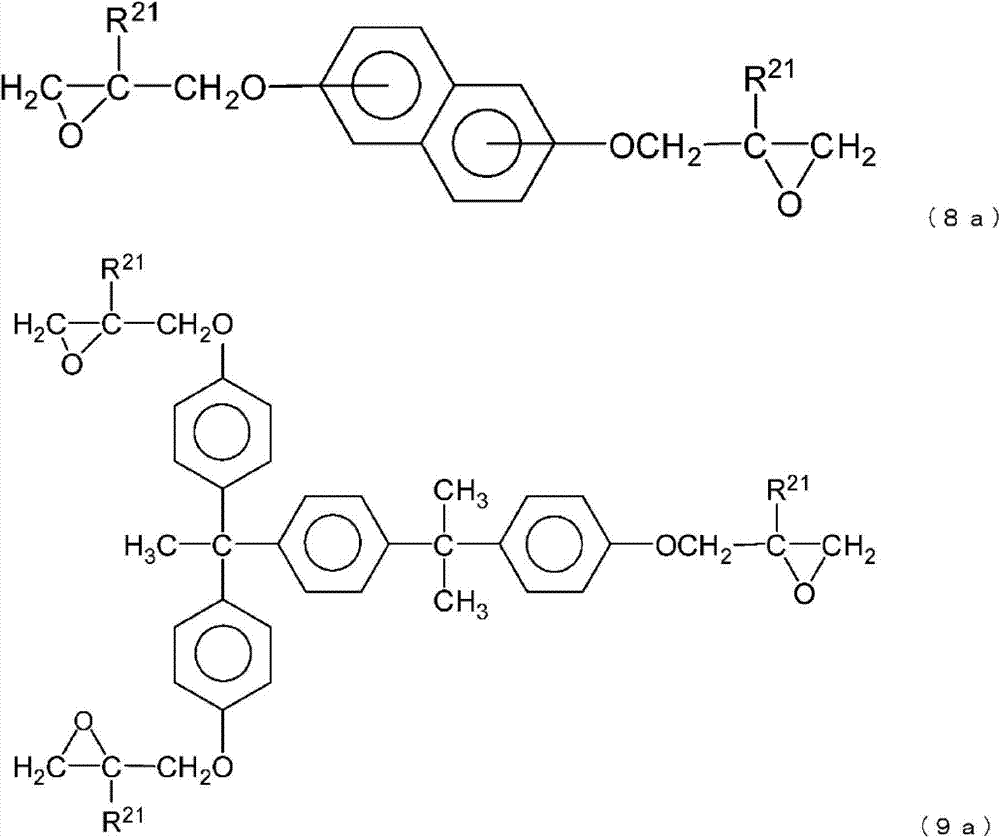
通式(7a)、通式(8a)和通式(9a)中,
X和R21如通式(1)中定义所述。
作为本发明的优选的多官能环氧化合物的、由通式(7a)~通式(9a)表示的环氧化合物,作为市售品例如可列举出EPICLON 850(DAINIPPONINK&CHEMICALS,INC制造)、EPIKOTE 828EL、EPIKOTE 1004(均为JAPAN EPOXY RESIN CO.,LTD.制造)等双酚A型环氧树脂,EPIKOTE806、EPIKOTE 4004(均为JAPAN EPOXY RESIN CO.,LTD.制造)等双酚F型环氧树脂,EPICLON HP4032、EPICLON EXA-4700(均为DAINIPPON INK & CHEMICALS,INC制造)等萘型环氧树脂,VG-3101(三井石油化学公司制造)等3官能环氧树脂。
在本发明中,分子中具有2个以上的羟基的多羟基化合物只要是分子中含有2个以上的羟基的化合物,就无特殊限定。作为多羟基化合物的具体例,可列举出以下化合物。
作为分子中具有2个羟基的化合物,可列举出乙二醇、二甘醇、三甘醇、丙二醇、一缩二丙二醇、新戊二醇、1,4-丁二醇和1,6-己二醇等单烷撑二醇和多烷撑二醇,邻苯二酚、1,2-二羟基萘、2,3-二羟基萘、1,2-二羟基蒽醌和2,3-二羟基喹喔啉等2元的芳香族羟基化合物,苯-1,4-二甲醇、苯-1,3-二甲醇、苯-1,4-二乙醇等芳香族醇,4-羟甲基苯酚、3-羟甲基苯酚、4-羟乙基苯酚和3-羟乙基苯酚等羟基烷基苯酚。
作为分子中具有3个羟基的化合物,可列举出甘油、三羟甲基丙烷、2-甲基-2-羟甲基-1,3-丙二醇、2,4-二羟基-3-羟甲基戊烷、1,2,6-己烷三醇和1,1,1-三(羟甲基)丙烷等3元醇,连苯三酚、3,4,5-三羟基甲苯、1,2,4-三羟基蒽醌、没食子酸以及没食子酸甲酯、没食子酸丙酯和没食子酸辛酯等没食子酸酯化合物等3元的芳香族多羟基化合物。
作为分子中具有4个以上的羟基的化合物,可列举出季戊四醇、一缩二甘油、四羟甲基甲烷、烷撑苷(甲基苷、乙基苷等)。另外,可列举出2,3,4,4’-四羟基二苯甲酮、鞣花酸、六羟基苯、鞣酸、和邻苯二酚或连苯三酚的杯芳烃化合物等4元以上的芳香族多羟基化合物。
在本发明中,多羟基化合物优选分子中具有2~6个的羟基的多羟基化合物,更优选分子中具有2~4个的羟基的多羟基化合物,特别优选为由下述通式(10)表示的二羟基化合物,
HO-Y-OH (10)
通式(10)中,Y如通式(1)中定义所述。
作为由通式(10)表示的二羟基化合物,可优选列举出乙二醇、丙二醇、丁烷-1,4-二醇二甘醇、三甘醇等单烷撑二醇和多烷撑二醇,苯-1,4-二甲醇、苯-1,3-二甲醇、苯-1,4-二乙醇等芳香族醇,(2-羟基苯基)甲醇、(2-羟基苯基)-2-乙醇等羟基烷撑基苯酚。
金属催化剂只要是用于环氧基的开环反应的催化剂,就可任选使用,例如可列举出由铜、锌、铁、镁、银、钙、锡等金属和BF4 -、SiF6 2-或PF6 -、CF3SO2 -等阴离子构成的金属催化剂。优选为氟硼酸亚锡(Sn(BF4)2)。
在本发明中,相对于1当量的多功能环氧化合物中的环氧基,多羟基化合物的用量为1当量~10当量,优选为4当量~8当量。在本发明中,可采用高效液相色谱法(HPLC),通过原料中的多官能环氧化合物以及单末端反应物的峰的消失,从而确认所有的环氧基与多羟基化合物发生反应而生成环氧开环体。在这里,所谓单末端反应物是指多官能环氧化合物的全部环氧基未开环的反应物,例如是在由通式(7a)和(8a)表示的化合物中仅1个环氧基发生开环的化合物,以及在由通式(9a)表示的化合物中仅1个或2个环氧基发生开环的化合物。
在本发明中,金属催化剂相对于全部反应混合物的重量而言为10~1,000ppm,优选为20~200ppm。
工序(1A)中的反应温度虽然无特殊限制,但为50℃~130℃、优选70℃~120℃。工序(1A)中的反应可在有机溶剂的存在下或没有有机溶剂的存在下进行。作为可使用的有机溶剂,可列举出如苯和甲苯之类的芳香族烃,例如环己酮之类的环式脂肪族酮,以及起始原料中的二羟基化合物。
工序(1B)
通过工序(1B),使在工序(1A)中得到的多官能环氧化合物的环氧开环体的羟基发生环氧化。在工序(1B)中,多官能环氧化合物的环氧开环体中的羟基的一部分或全部发生环氧化。在本发明中,优选多官能环氧化合物的环氧开环体的羟基的50%~100%发生环氧化,更优选75%~100%发生环氧化。
多官能环氧化合物的环氧开环体是工序(1B)中的原料化合物,可列举出上述多官能环氧化合物的环氧基全部开环而得的化合物。优选作为由通式(7a)~通式(9a)表示的环氧化合物的环氧开环体的、由下述通式(7b)、通式(8b)或通式(9b)表示的多官能环氧化合物的环氧开环体,


式中,X、Y和R21如上述定义所述。
在工序(1B)中,环氧化可采用公知的将羟基环氧化的反应,例如可列举出表氯醇法和氧化法,优选为表氯醇法。
表氯醇法是一种通过使在工序(1A)中得到的多官能环氧化合物的环氧开环体在相间转移催化剂的存在下与表氯醇或甲基表氯醇发生反应,从而将多官能环氧化合物的环氧开环体的羟基环氧化的方法。
在表氯醇法中,可以以达到所需的环氧基数量的摩尔数而使表氯醇或甲基表氯醇反应。相对于1摩尔的多官能环氧化合物的环氧开环体的羟基,表氯醇或甲基表氯醇的量为0.5~5摩尔、优选为0.5~2.5。例如,相对于1摩尔的分子中具有4个羟基的由通式(7b)和通式(8b)表示的化合物,表氯醇或甲基表氯醇的量为2~20摩尔、优选2~10摩尔。相对于1摩尔的分子中具有6个羟基的由通式(9b)表示的化合物,表氯醇或甲基表氯醇的量为3~30摩尔、优选3~15摩尔。
相间转移催化剂可列举出如甲基三辛基氯化铵、甲基三癸基氯化铵和四甲基氯化铵之类的四烷基氯化铵,以及如苄基三甲基氯化铵之类的芳烷基三烷基氯化铵等季铵盐,优选苄基三甲基氯化铵。相间转移催化剂的用量基于反应体的总重量而言为0.1~5重量%,更优选为0.5~2.0重量%。
反应可在如己烷和戊烷等烃,二乙醚、叔丁基甲基醚和二异丙醚等醚,或丙酮和甲乙酮等酮之类的溶剂的存在下进行,但作为溶剂也可使用过量的表氯醇和甲基表氯醇。反应温度为30~90℃、优选40~65℃,最优选可在约50至约55℃范围的温度下反应。
氧化法是一种包括以下工序的方法,即将在工序(1A)中得到的多官能环氧化合物的环氧开环体的羟基加以烯丙基化,得到二烯丙基醚化合物的工序,和将二烯丙基醚化合物的烯丙基或2-甲基-2-丙烯基加以氧化的工序。在本发明中,羟基的烯丙基化包括使羟基转化为烯丙基或2-甲基-2-丙烯基。
在本发明中,得到二烯丙基醚化合物的工序是一种通过使多官能环氧化合物的环氧开环体与烯丙基卤化物或2-甲基-2-丙烯基卤化物反应,从而将多官能环氧化合物的环氧开环体的羟基转化为烯丙基或2-甲基-2-丙烯基的工序。具体而言,在将多官能环氧化合物的环氧开环体和烯丙基卤化物溶于二甲基亚砜后,添加季铵盐,在将反应温度保持在40℃以下的同时滴加碱水溶液,在滴加结束后,于30~40℃进行约6小时的反应。
作为烯丙基卤化物和2-甲基-2-丙烯基卤化物中的卤素,可列举出氯和溴。相对于1摩尔的多官能环氧化合物的环氧开环体的羟基,烯丙基卤化物和2-甲基-2-丙烯基卤化物的添加量优选为3~30摩尔。
作为季铵盐,例如可列举出四丁基溴化铵等四烷基卤化铵或四苯基氯化铵等四芳基卤化铵。相对于1摩尔的多官能环氧化合物的环氧开环体,季铵盐的添加量优选为0.001摩尔~0.1摩尔。
作为碱水溶液,可列举出氢氧化钙、氢氧化钾和氢氧化钠。相对于1当量的多官能环氧化合物的环氧开环体的羟基,所使用的碱金属的用量优选为2~8当量。
对二烯丙基醚化合物的烯丙基或2-甲基-2-丙烯基进行氧化的工序是一种使二烯丙基醚化合物在碳酸钾的存在下与过氧化氢水发生反应的工序。具体而言,在将多官能环氧化合物的环氧开环体的羟基加以烯丙基化而得的二烯丙基醚化合物中,添加甲醇、乙醇等醇或者乙腈、苄腈等腈等溶剂和碳酸钾,在搅拌下滴加5~40%、优选30~35%的过氧化氢水,在滴加结束后,进行0.5~10小时、优选1~6小时的氧化反应。
过氧化氢水的添加量优选相对于1摩尔的将多官能环氧化合物的环氧开环体的羟基加以烯丙基化而得的二烯丙基醚化合物而言添加至5~15摩尔。反应温度例如为45℃以下、优选20~40℃。
本发明的环氧树脂的制造方法优选为本发明的由通式(1)~通式(3)表示的环氧树脂的制造方法,所述环氧树脂的制造方法包括下述工序(2A)~(2B):
(2A)通过使由通式(7a)、通式(8a)或通式(9a)表示的环氧化合物在金属催化剂的存在下与由下述通式(10)表示的二羟基化合物反应,从而得到由下述通式(7b)、通式(8b)或通式(9b)表示的环氧开环体的工序,

通式(7a)、通式(8a)和通式(9a)中,
X和R21如通式(1)中定义所述,
HO-Y-OH (10)
通式(10)中,Y如通式(1)中定义所述,
通式(7b)、通式(8b)和通式(9b)中,
X、Y和R21如上述定义所述;和
(2B)使在工序(2A)中得到的由通式(7b)~通式(9b)表示的环氧开环体的羟基发生环氧化,得到由通式(1)~通式(3)表示的环氧树脂的工序。
部分酯化环氧树脂的制造方法
接着,对本发明的部分酯化环氧树脂的制造方法进行说明。本发明的部分酯化环氧树脂的制造方法包括工序(1C):
(1C)使通过包括上述工序(1A)~(1B)的制造方法制得的环氧树脂在碱性催化剂的存在下与(甲基)丙烯酸发生反应的工序。
工序(1C)
在工序(1C)中,使通过包括上述工序(1A)~(1B)的制造方法制得的环氧树脂的缩水甘油基和甲基缩水甘油基发生(甲基)丙烯酰基化。作为通过包括工序(1A)~(1B)的制造方法制得的环氧树脂,优选为由通式(1)~通式(3)表示的环氧树脂。
作为(甲基)丙烯酸,无特殊限定,例如可使用市售的丙烯酸或甲基丙烯酸。
在本发明中,在使通过包括工序(1A)~(1B)的制造方法制得的环氧树脂与(甲基)丙烯酸发生反应的工序中,相对于1当量的通过包括工序(1A)~(1B)的制造方法制得的环氧树脂的环氧基而言,发生反应的(甲基)丙烯酸优选为10~90当量%,更优选为20~80当量%,进一步优选为30~70当量%,特别优选为40~60当量%。在部分酯化环氧树脂的制造方法中,由于缩水甘油基和甲基缩水甘油基与(甲基)丙烯酸的反应定量地进行,所以得到的部分酯化环氧树脂的酯化率也可根据环氧当量来推测。
相对于1当量的通过包括工序(1A)~(1B)的制造方法制得的环氧树脂的环氧基,若在上述范围内使(甲基)丙烯酸发生反应,则可得到部分酯化环氧树脂,该部分酯化环氧树脂是在仅使不饱和基团发生反应的一次聚合时能够暂时固定地获得良好的树脂特性,在二次聚合时能够不产生相分离等就形成均匀的聚合物的部分酯化环氧树脂。
作为碱性催化剂,可使用基于环氧树脂与(甲基)丙烯酸的反应所使用的公知的碱性催化剂。另外,也可使用将碱性催化剂搭载于聚合物而得的聚合物担载型碱性催化剂。
作为碱性催化剂,优选为3价的有机磷化合物和/或胺化合物。碱性催化剂的碱性原子为磷和/或氮。
作为3价的有机磷化合物,可列举出如三乙基膦、三正丙基膦、三正丁基膦之类的烷基膦类及其盐,三苯基膦、三间甲苯基膦、三-(2,6-二甲氧基苯基)膦等芳基膦类及其盐,亚磷酸三苯酯、亚磷酸三乙酯、亚磷酸三(壬基苯酯)等亚磷酸三酯类及其盐等。作为3价的有机磷化合物的盐,可列举出乙基三苯基溴化膦、丁基三苯基溴化膦、辛基三苯基溴化膦、癸基三苯基溴化膦、异丁基三苯基溴化膦、丙基三苯基氯化膦、戊基三苯基氯化膦、己基三苯基溴化膦等。其中,优选为三苯基膦。
作为胺化合物,可列举出二乙醇胺等仲胺,三乙醇胺、二甲基苄胺、三(二甲基氨基甲基)苯酚、三(二乙基氨基甲基)苯酚等叔胺,1,5,7-三氮杂双环[4.4.0]癸-5-烯(TBD)、7-甲基-1,5,7-三氮杂双环[4.4.0]癸-5-烯(Me-TBD)、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)、6-二丁基氨基-1,8-二氮杂双环[5.4.0]十一碳-7-烯、1,5-二氮杂双环[4.3.0]壬-5-烯(DBN)、1,1,3,3-四甲基胍等强碱性胺及其盐。其中,优选1,5,7-三氮杂双环[4.4.0]癸-5-烯(TBD)。作为胺化合物的盐,可列举出苄基三甲基氯化铵、苄基三乙基氯化铵。
作为担载碱性催化剂的聚合物,无特殊限定,可使用通过二乙烯基苯使聚苯乙烯交联而得的聚合物或通过二乙烯基苯使丙烯酸树脂交联而得的聚合物等。这些聚合物不溶于在通过包括工序(1A)~(1B)的制造方法制得的环氧树脂与(甲基)丙烯酸的反应中所使用的溶剂(例如甲乙酮、甲基异丁基酮、甲苯等)和原料、生成物中。
聚合物担载型碱性催化剂通过使碱性催化剂与不溶性聚合物发生化学键合,或在将碱性催化剂导入单体后将单体聚合然后通过二乙烯基苯等交联单体三维地进行交联,从而可制造不溶于甲乙酮、甲基异丁基酮、甲苯等溶剂的聚合物担载型碱性催化剂。
作为聚合物担载型碱性催化剂,具体而言,可列举出二苯基膦基聚苯乙烯、1,5,7-三氮杂双环[4.4.0]癸-5-烯聚苯乙烯、N,N-(二异丙基)氨基甲基聚苯乙烯、N-(甲基聚苯乙烯)-4-(甲基氨基)吡啶等。这些聚合物担载型碱性催化剂可单独使用或合用2种以上使用。
聚合物担载型碱性催化剂可使用市售的产品。作为市售的聚合物担载型碱性催化剂,例如可列举出PS-PPh3(二苯基膦基聚苯乙烯,BIOTAGEJAPAN LTD.制造)、PS-TBD(1,5,7-三氮杂双环[4.4.0]癸-5-烯聚苯乙烯,BIOTAGE JAPAN LTD.制造)。
聚合物担载型碱性催化剂的使用比例为,相对于1当量的通过包括工序(1A)~(1B)的制造方法制得的环氧树脂的环氧基而言,聚合物担载型碱性催化剂优选为0.5~5.0毫当量,更优选为1.0~3.0毫当量。若聚合物担载型碱性催化剂的使用比例在上述范围内,则从反应率、反应时间和催化剂成本的观点出发而优选。
在本发明的制造方法中,通过包括工序(1A)~(1B)的制造方法得到的环氧树脂与(甲基)丙烯酸的反应工序中的温度优选为60~120℃,更优选为80~120℃,进一步优选为90~110℃。
在催化剂存在下使通过包括工序(1A)~(1B)的制造方法得到的环氧树脂与(甲基)丙烯酸发生反应的情况下,为防止凝胶化,需要适当保证反应体系内和反应体系上的气相的氧浓度。例如,由于在积极地向反应体系内吹入空气的情况下有时会引起催化剂的氧化而导致活性的降低,所以需要注意。
就通过包括工序(1A)~(1B)的制造方法得到的环氧树脂与(甲基)丙烯酸的反应而言,由于通过此反应而得的部分酯化环氧树脂因紫外线等活性能量射线而发生固化,所以希望在遮蔽紫外线的容器内进行反应。另外,为防止气相聚合,通过包括工序(1A)~(1B)的制造方法得到的环氧树脂与(甲基)丙烯酸的反应也可在对环氧树脂呈现出良溶剂性的回流溶剂存在下进行,但是由于在此情况下需要在反应结束后除去溶剂,所以优选在无溶剂下进行。作为回流溶剂,可列举出丙酮、甲乙酮等。
在本发明的制造方法中,在使通过包括工序(1A)~(1B)的制造方法得到的环氧树脂与(甲基)丙烯酸反应后,通过除去聚合物担载型碱性催化剂而得到部分酯化环氧树脂。作为除去聚合物担载型碱性催化剂的方法,优选采用过滤或离心分离。
作为过滤聚合物担载型碱性催化剂的方法,例如可列举出使用孔径为10μm的尼龙筛NY-10HC(瑞士Sefar公司制造)来过滤出聚合物担载型碱性催化剂的方法。
作为将聚合物担载型碱性催化剂加以离心分离的方法,可列举出通过使用离心分离机进行固液分离,从而除去聚合物担载型碱性催化剂的方法。
由此,本发明的部分酯化环氧树脂的制造方法优选为上述所述的部分酯化环氧树脂的制造方法,其是用于制造由通式(4)~通式(6)表示的部分酯化环氧树脂的方法,包括工序(2C):
(2C)使通过包括工序(2A)~(2B)的工序的制造方法而得的由通式(1)、通式(2)或通式(3)表示的环氧树脂在碱性催化剂的存在下与(甲基)丙烯酸发生反应,得到由通式(4)~通式(6)表示的部分酯化环氧树脂的工序。
固化性组合物
对本发明的固化性组合物进行说明,所述固化性树脂含有选自(a)由通式(1)、通式(2)和通式(3)表示的环氧树脂,(b)由通式(4)、通式(5)和通式(6)表示的部分酯化环氧树脂,(c)通过包括工序(1A)~(1B)的制造方法而得的环氧树脂,以及(d)通过包括工序(1C)的制造方法而得的部分酯化环氧树脂中的1种以上的树脂。在本发明中,作为固化性树脂中含有的基质低聚物成分的树脂是选自本发明的环氧树脂和本发明的部分酯化环氧树脂中的1种以上的树脂。例如,可将由通式(1)、通式(2)或通式(3)表示的环氧树脂或者由通式(4)、通式(5)或通式(6)表示的部分酯化环氧树脂单独使用,或者可将由通式(1)、通式(2)或通式(3)表示的环氧树脂或者由通式(4)、通式(5)或通式(6)表示的部分酯化环氧树脂的2种以上混合使用。
作为本发明的固化性组合物中含有的成分,除了本发明的环氧树脂和部分酯化环氧树脂以外,还可列举出固化剂、聚合引发剂、填充剂、偶联剂。
作为固化剂,无特殊限定,可使用作为固化剂而公知的化合物。作为固化剂,可列举出胺系固化剂,例如有机酸二酰肼化合物、咪唑及其衍生物、双氰胺、芳香族胺、环氧改性多胺和聚氨基脲等,特别优选作为有机酸二酰肼的VDH(1,3-双(肼基羰基乙基)-5-异丙基乙内酰脲)、ADH(己二酸二酰肼)、UDH(7,11-十八烷二烯-1,18-双(肼基羰基))和LDH(十八烷-1,18-二甲酸二酰肼)。这些固化剂可单独使用或合用多种。相对于100重量份的环氧树脂和部分酯化环氧树脂而言,引发剂的配合量优选为1~25重量份,更优选为5~15重量份。
聚合引发剂是指通过吸收光的能量而进行活化,从而产生自由基的化合物。聚合引发剂无特殊限定,可使用作为聚合引发剂而公知的化合物。作为聚合引发剂,可列举出苯偶姻类、苯乙酮类、二苯甲酮类、噻吨酮类、α-酰基肟酯类(α-アシロキシムエステル類)、乙醛酸苯酯类(フエニルグリオキシレ一ト類)、偶苯酰类、偶氮系化合物、二苯硫醚系化合物、酰基氧化膦系化合物、苯偶姻类、苯偶姻醚类和蒽醌类聚合引发剂,优选为在液晶中的溶解性低,而且其自身具有在光照射时分解物不发生气化的反应性基团的聚合引发剂。作为本发明的优选的聚合引发剂,例如可列举出EY树脂KR-2(KSM CO.,LTD.制造)等。相对于100重量份的环氧树脂和部分酯化环氧树脂而言,聚合引发剂的配合量优选为0.1~5重量份,更优选为1~5重量份。
填充剂以控制固化性组合物的粘度或提高使固化性组合物固化而得的固化物的强度,或通过抑制直线膨胀性而提高固化性组合物的粘结可靠性等为目的而添加。填充剂可使用对于含有环氧树脂的组合物所使用的公知的无机填充剂和有机填充剂。作为无机填充剂,可列举出碳酸钙、碳酸镁、硫酸钡、硫酸镁、硅酸铝、氧化钛、氧化铝、氧化锌、二氧化硅、高岭土、滑石、玻璃珠、绢云母活性粘土、膨润土、氮化铝和氮化硅。作为有机填充剂,可列举出聚甲基丙烯酸甲酯、聚苯乙烯、使构成它们的单体与其它的单体共聚而得的共聚物、聚酯微粒、聚氨酯微粒和橡胶微粒。在本发明中,特别优选无机填充剂,例如二氧化硅和滑石。相对于100重量份的环氧树脂和部分酯化环氧树脂而言,填充剂的配合量优选为2~40重量份,更优选为5~30重量份。
偶联剂以使与液晶显示基板的粘结性更加良好为目的而添加。作为偶联剂,无特殊限定,例如可列举出γ-氨基丙基三甲氧基硅烷、γ-巯基丙基三甲氧基硅烷、γ-异氰酸酯基丙基三甲氧基硅烷和3-环氧丙氧基丙基三甲氧基硅烷等。这些硅烷偶联剂可单独使用或合用2种以上使用。相对于100重量份的环氧树脂和部分酯化环氧树脂而言,硅烷偶联剂的配合量优选为0.1~10重量份,更优选为0.5~2重量份。
本发明的固化性组合物通过照射紫外线等能量射线、通过加热或通过在照射紫外线等能量射线后加热而固化。由此可得到本发明的固化性组合物的固化物。因此,使含有本发明的环氧树脂和部分酯化环氧树脂的固化性组合物进行固化的方法包括:对含有本发明的环氧树脂和部分酯化环氧树脂的固化性组合物照射紫外线等能量射线,进行加热,或在照射紫外线等能量射线后进行加热的工序。
本发明的环氧树脂和部分酯化环氧树脂在液晶中的溶出性极低。另外,就使用了含有本发明的环氧树脂和部分酯化环氧树脂的固化性组合物的液晶显示元件而言,即使在UV照射量低的情况下,液晶的取向性仍良好,没有产生显示不良,因此,可用作液晶用密封剂。
实施例
接下来,通过实施例对本发明的具体实施方式进行更详细的说明,但本发明不受这些实施例的限定。
[合成例1]化合物1c的制造
(1-1)化合物1a(双酚A型环氧树脂的乙二醇开环体,是在通式(7b)中X为异丙叉基、X的键合位置为4,4’-位、R21均为氢、Y为乙撑基的化合物)的合成
将500g的乙二醇(东京化成公司制造)、1.0g的45%氟硼酸亚锡(II)水溶液(森田化学工业公司制造)加入茄形烧瓶中。在搅拌340g的双酚A型环氧树脂(EXA850CRP,DIC株式会社制造)的同时将其保持在80℃,用1小时缓慢加入,从添加结束起在80℃下搅拌1小时。将反应混合物冷却至室温,加入1L的二氯甲烷,用1L的水洗涤6次。通过减压蒸馏除去所得的有机相的溶剂,得到410g的无色透明粘稠物的开环体(化合物1a)。需要说明的是,通过高效液相色谱法(HPLC),根据原料中的环氧树脂和仅1个环氧基发生了开环的化合物的峰的消失,从而确认出所有环氧基均发生开环。
(1-2)化合物1b(在通式(1)中,X为异丙叉基,X的键合位置为4,4’-位,R21均为氢,Y为乙撑基,R1、R2、R4和R5之中3.8个为缩水甘油基的化合物)的合成
将232g的化合物1a(EXA850CRP-乙二醇开环体)、590g的表氯醇(和光纯药公司制造)、50g的苄基三甲基氯化铵(东京化成公司制造)加入到安装有机械搅拌器、温度计、温度调节器、冷凝器、Dean-Stark分水器和滴液漏斗的2升的三颈圆底烧瓶中。接着,将混合物在70托(torr)的高真空下搅拌的同时加热至约50至55℃,使表氯醇剧烈回流。将160g的48%溶液NaOH(关东化学公司制造)用2小时缓慢添加到混合物中。生成共沸物后,在水/表氯醇混合物之中将表氯醇送回反应体系,与此同时继续搅拌。在添加结束后,继续搅拌3小时。接着,将反应混合物冷却至室温,加入1L的二氯甲烷,用1L的水洗涤6次。通过减压蒸馏而除去得到的有机相的溶剂,得到245g的淡黄色透明粘稠物的缩水甘油醚体(化合物1b)。根据HPLC,分子中的环氧基为3.8个。
(1-3)化合物1c(是在通式(4)中,X为异丙叉基,X的键合位置为4,4’-位,R21均为氢,Y为乙撑基,R11、R12、R14和R15之中1.9个为缩水甘油基和甲基丙烯酰基,缩水甘油基与甲基丙烯酰基的比例为50∶50的化合物)的合成
将210g的化合物1b、88.7g的甲基丙烯酸(东京化成公司制造)、4.7g的PS-PPh3(BIOTAGE JAPAN LTD.制造)、10.3g的PS-TBD(BIOTAGEJAPAN LTD.制造)、100mg的BHT混合,于100℃搅拌6小时。在反应结束后,通过过滤除去催化剂,得到部分甲基丙烯酸酯化环氧树脂。
[合成例2]化合物2c的制造
(2-1)化合物2a(双酚A型环氧树脂的2-(4-羟基苯基)乙醇开环体,是在通式(7b)中X为异丙叉基、X的键合位置为4,4’-位、R21为氢、Y为亚甲基-亚苯基的化合物)的合成
将170g的双酚A型环氧树脂(EXA850CRP,DIC株式会社制造)、138g的2-(4-羟基苯基)乙醇(东京化成公司制造)、2.3g的PS-PPh3(BIOTAGE JAPAN LTD.制造)、4.2g的PS-TBD(BIOTAGE JAPAN LTD.制造)、500ml的甲苯加入到茄形烧瓶中,于100℃搅拌24小时。将反应液冷却至室温,通过过滤而除去催化剂。通过减压蒸馏而除去得到的混合物的溶剂,得到285g的淡黄色透明粘稠物的开环体。
(2-2)化合物2b(是在通式(1)中X为异丙叉基,X的键合位置为4,4’-位,R21均为氢,Y为亚甲基-亚苯基,R1、R2、R4和R5之中3.6个为缩水甘油基的化合物)的合成
将185g的化合物2a(EXA850CRP-2-(4-羟基苯基)乙醇开环体)、590g的表氯醇(和光纯药公司制造)、22g的苄基三甲基氯化铵(东京化成公司制造)加入到安装有机械搅拌器、温度计、温度调节器、冷凝器、Dean-Stark分水器和滴液漏斗的2L三颈圆底烧瓶中。接着,将混合物在70托(torr)的高真空下搅拌的同时将其加热至约50至55℃,使表氯醇剧烈回流。将185g的48%溶液NaOH(关东化学公司制造)用2小时缓慢添加到混合物中。生成共沸物后,在水/表氯醇混合物之中,在将表氯醇送回反应体系的同时继续搅拌。在添加结束后,继续搅拌4小时。接着,将反应混合物冷却至室温,加入1L的二氯甲烷,用1L的水洗涤6次。通过减压蒸馏而除去得到的有机相的溶剂,得到233g的黄色透明粘稠物的缩水甘油醚体(化合物2b)。根据HPLC,分子中的环氧基为3.6个。
(2-3)化合物2c(是在通式(4)中X为异丙叉基,X的键合位置为4,4’-位,R21均为氢,Y为亚甲基-亚苯基,R1、R2、R4和R5之中1.8个为缩水甘油基和甲基丙烯酰基,缩水甘油基与甲基丙烯酰基的比例为50∶50的化合物)的合成
将180g的化合物2b、36g的甲基丙烯酸(东京化成公司制造)、1.9g的PS-PPh3(BIOTAGE JAPAN LTD.制造)、8.3g的PS-TBD(BIOTAGEJAPAN LTD.制造)、85mg的BHT(和光纯药公司制造)混合,于100℃搅拌6小时。在反应结束后,通过过滤而除去催化剂,得到部分甲基丙烯酸酯化环氧树脂。
[合成例3]化合物3c的制造
(3-1)化合物3a(萘型环氧树脂的乙二醇开环体,是在通式(8b)中R21为氢、Y为乙撑基、萘环的键合位置为1,6-位的化合物)的合成
将500g的乙二醇(东京化成公司制造)、1.0g的45%氟硼酸亚锡(II)水溶液(森田化学工业公司制造)加入茄形烧瓶中。将萘型环氧树脂(HP-4032D,DIC株式会社制造)282g在搅拌的同时保持在100℃,用1小时缓慢加入,从添加结束起于100℃搅拌3小时。将反应混合物冷却至室温,加入1L的二氯甲烷,用1L的水洗涤6次。通过减压蒸馏而除去得到的有机相的溶剂,得到390g的黄色透明粘稠物的开环体(化合物3a)。
(3-2)化合物3b(是在通式(2)中R21均为氢,Y为乙撑基,萘环的键合位置为1,6-位,R1、R2、R4和R5之中3.9个为缩水甘油基的化合物)的合成
将112g的化合物3a(HP4032D-乙二醇开环体)、590g的表氯醇(和光纯药公司制造)、21g的苄基三甲基氯化铵(东京化成公司制造)加入到安装有机械搅拌器、温度计、温度调节器、冷凝器、Dean-Stark分水器和滴液漏斗的2升的三颈圆底烧瓶中。接着,将混合物在70托(torr)高真空下搅拌,与此同时加热至约50至55℃,使表氯醇剧烈回流。将188g的48%溶液NaOH(关东化学公司制造)经2小时缓慢添加到混合物中。生成共沸物后,在水/表氯醇混合物之中,在将表氯醇送回反应体系的同时继续搅拌。在添加结束后,继续搅拌4小时。接着,将反应混合物冷却至室温,加入1L的二氯甲烷,用1L的水洗涤6次。通过减压蒸馏而除去得到的有机相的溶剂,得到179g的淡黄色透明粘稠物的缩水甘油醚体(化合物3b)。根据HPLC,分子中的环氧基为3.9个。
(3-3)化合物3c(是在通式(5)中R21均为氢,Y为乙撑基,萘环的键合位置为1,6-位,R11、R12、R14和R15之中1.9个为缩水甘油基和甲基丙烯酰基,缩水甘油基与甲基丙烯酰基的比例为50∶50的化合物)的合成
将50g的化合物3b、11.8g的甲基丙烯酸(东京化成公司制造)、154mg的PS-PPh3(BIOTAGE JAPAN LTD.制造)、341mg的PS-TBD(BIOTAGE JAPAN LTD.制造)、12mg的BHT混合,于100℃搅拌6小时。在反应结束后,通过过滤而除去催化剂,得到部分甲基丙烯酸酯化环氧树脂(化合物3c)。
[合成例4]化合物4c的制造
(4-1)化合物4a(VG3101L的丙二醇开环体,是在通式(9b)中R21均为氢、Y为亚丙基的化合物)的合成
将148g的TECHMORE VG3101L(PRINTEC CORPORATION制造)、456g的丙二醇(关东化学公司制造)、130mg的45%氟硼酸亚锡(II)水溶液(森田化学工业公司制造)加入茄形烧瓶中,于100℃搅拌20小时。将反应混合物冷却至室温,加入1L的二氯甲烷,用1L的水洗涤6次。通过减压蒸馏而除去得到的有机相的溶剂,得到190g的黄色粘稠物的开环体。
(4-2)化合物4b(是在通式(3)中R21均为氢,Y为亚丙基,R1~R6之中5.8个为缩水甘油基的化合物)的合成
将160g的化合物4a(VG3101L-丙二醇开环体)、590g的表氯醇(和光纯药公司制造)、22g的苄基三甲基氯化铵(东京化成公司制造)加入到安装有机械搅拌器、温度计、温度调节器、冷凝器、Dean-Stark分水器和滴液漏斗的2升的三颈圆底烧瓶中。接着,将混合物在70托(torr)的高真空下搅拌的同时加热至约50至55℃,使表氯醇剧烈回流。将117g的48%溶液NaOH(关东化学公司制造)用2小时缓慢添加到混合物中。生成共沸物后,在水/表氯醇混合物之中,在将表氯醇送回反应体系的同时继续搅拌。在添加结束后,继续搅拌4小时。接着,将反应混合物冷却至室温,加入1L的二氯甲烷,用1L的水洗涤6次。通过减压蒸馏而除去得到的有机相的溶剂,得到207g的黄色粘稠物的缩水甘油醚体(化合物4b)。根据HPLC,分子中的环氧基为5.8个。
(4-3)化合物4c(是在通式(6)中R21均为氢,Y为亚丙基,R11R16之中5.8个为缩水甘油基和甲基丙烯酰基,缩水甘油基与甲基丙烯酰基的比例为75∶25的化合物)的合成
将34g的化合物4b、3.2g的甲基丙烯酸(东京化成公司制造)、41mg的PS-PPh3(BIOTAGE JAPAN LTD.制造)、75mg的PS-TBD(BIOTAGEJAPAN LTD.制造)、10mg的BHT混合,于100℃下搅拌6小时。在反应结束后,通过过滤而除去催化剂,得到部分甲基丙烯酸酯化环氧树脂。
[比较合成例1]
(5-1)比较化合物5c(合成例1中使用的双酚A型环氧树脂的部分甲基丙烯酸酯化环氧树脂)的合成
将320.2g的双酚A型环氧树脂(EXA850CRP,DIC株式会社制造)、90.4g的甲基丙烯酸(东京化成公司制造)、1.5g的PS-PPh3(BIOTAGEJAPAN LTD.制造)、100mg的BHT混合,于100℃搅拌6小时。在反应结束后,通过过滤而除去催化剂,得到部分甲基丙烯酸酯化环氧树脂(比较化合物5c)。
将合成例1~4中得到的环氧开环体、环氧树脂和部分(甲基)丙烯酸酯化环氧树脂的、使用E型粘度计(东机产业公司制造RE105U)在圆锥形转子的转速为2.5rpm下测定粘度而得的粘度,根据JIS K 7236:2001测得的环氧当量和丙烯酰化率示于表1中。在这里,丙烯酰化率根据所使用的环氧当量和甲基丙烯酸当量的当量比算出。
[表1]
| 化合物 | 1a | 1b | 1c | 2a | 2b | 2c |
| 粘度(mPa·s) | 粘稠物 | 27,600 | 209,400 | 粘稠物 | 94,800 | 240,000 |
| 环氧当量(g/eq.) | - | 206 | 590 | - | 220 | 490 |
| 丙烯酰化率(%) | - | - | 50 | - | - | 50 |
| 化合物 | 3a | 3b | 3c | 4a | 4b | 4c |
| 粘度(mPa·s) | 粘稠物 | 15,000 | 121,200 | 粘稠物 | 212,400 | 559,200 |
| 环氧当量(g/eq.) | - | 181 | 420 | - | 225 | 280 |
| 丙烯酰化率(%) | - | - | 50 | - | - | 25 |
针对合成例1~4中制造的环氧树脂(化合物1b、2b、3b和4b)和部分酯化环氧树脂(化合物1c、2c、3c和4c)以及合成例1中使用的作为原料化合物的双酚A型环氧树脂(比较化合物1a)、合成例3中使用的作为原料化合物的萘型环氧树脂(比较化合物3a)、合成例4中使用的作为原料化合物的3官能型环氧树脂(TECHMORE VG3101L(PRINTECCORPORATION制造),比较化合物4a)和比较合成例1中制造的双酚A型环氧树脂的部分甲基丙烯酸酯化环氧树脂(比较化合物5c)的各种低聚物,如下地进行溶出性试验。
[溶出性试验]
在液晶中的溶出性的评价通过作为液晶的相变温度的Ni点(Nematic-Isotropic point)的变化和利用HPLC(高效液相色谱法)的在液晶中的溶出量的直接定量来进行。液晶的Ni点由液晶中的各种成分的混合组成来决定,在各种配合下为固有的值。一般而言,已知Ni点因这些在液晶中混入某些杂质(其它成分)而降低,可通过Ni点来评价杂质混入情况。[测定用试样的制作]
在安瓿中加入0.1g的低聚物,加入1g的液晶(MLC-11900-080,MERCK & CO.,INC.制造)。将此瓶投入120℃烘箱中1小时,此后于室温静置,在恢复至室温(25℃)后取出液晶部分,用0.2μm过滤器过滤,制成评价用液晶试样。
[Ni点的测定]
Ni点的测定使用差示扫描量热计(DSC,PERKIN ELMER INC.制造,Pyris6)。将10mg的评价用液晶试样封入到铝试样盘中,在升温速度为5℃/分钟的条件下进行测定。
[溶出量的计算]
液晶中的低聚物的溶解量使用HPLC进行测定。HPLC测定用试样使用将取出的液晶用乙腈稀释至100倍而得的试样。另外,溶出量的定量根据各化合物的峰面积的检量线算出。将用于各试验的化合物的粘度和各试验的结果示于表2和表3中。
[表2]

[表3]

接着,使用合成例1和2中制造的部分酯化环氧树脂(化合物1c和2c)以及比较合成例1中制造的部分酯化环氧树脂(比较化合物5c,双酚A型环氧树脂的部分甲基丙烯酸酯化环氧树脂),以如表4所示的配合比来制作树脂组合物,进行测试元件中的液晶取向性的评价。
[取向性评价]
在将合成例1和3以及比较合成例1中得到的化合物(化合物1c、3c和5c)分别与EY树脂KR-2(KSM CO.,LTD.制造)、SEAHOSTAR KE-C50HG(日本催化剂公司制造)、己二酸二酰肼(大冢化学公司制造)和KBM-403(硅烷偶联剂:信越化学工业制造)按如表4所示的配合量混合后,使用3联辊充分进行混炼,得到各种树脂组合物(实施例9、实施例10、和比较例5)。将这样得到的树脂组合物用密封滴下器在4000μm2的剖面积中滴下涂布在带有经研磨处理后的取向膜(SUNEVERSE-7492,日产化学工业公司制造)的ITO玻璃基板上(60mm×70mm×0.7mmt)。然后,在基板上滴下液晶(TN液晶,MLC-11900-080,MERCK & CO.,INC.制造),通过液晶滴下工艺(ODF工艺)使上下基板粘合,照射紫外线(UV照射装置:UVX-01224S1,USHIO INC.制造,照度和照射时间:在1000mJ的情况下100mW/cm2/365nm、10秒,在50mJ的情况下50mW/cm2/365nm、1秒)使之固化,然后在120℃的热风烘箱中进行1小时的热固化,制作用于取向性试验的测试元件。另外,在照度为0mJ的情况下,在粘合后用遮光掩模形成紫外线不照射液晶和密封剂的状态,在120℃的热风烘箱中进行1小时的热固化,制作用于取向性试验的测试元件。
对于得到的面板,进行密封时的液晶的取向状态的确认。确认通过光学显微镜进行,将偏振片以正交尼科尔棱镜(クロスニコル)的状态夹住测试元件,通过透射进行观察。将显示1000mJ、50mJ和0mJ照度下的液晶的拐角部和直线部的取向状态的显微镜照片示于图1~图6中。需说明的是,图1~图6均为纵900μm×横1200μm的比例尺。液晶的取向性的评价标准根据密封时有无取向紊乱来判断。将密封时取向紊乱为50μm以下的情况计为“○”,将有更大的取向紊乱的情况计为“×”。将结果示于表4中。需说明的是,表4中的含量均为重量份。
[表4]
| 配合(重量份) | 实施例9 | 实施例10 | 比较例5 |
| 化合物1c | 100 | ||
| 化合物3c | 100 | ||
| 比较化合物5c | 100 | ||
| 引发剂 | 2 | 2 | 2 |
| 固化剂 | 10 | 10 | 10 |
| 填充剂 | 10 | 10 | 10 |
| 偶联剂 | 1 | 1 | 1 |
| 取向性(0mJ) | ○ | ○ | × |
| 取向性(50mJ) | ○ | ○ | × |
| 取向性(1000mJ) | ○ | ○ | ○ |
| 溶出性(Ni点) | ○ | ○ | × |
引发剂:EY树脂KR-2(KSM CO.,LTD.制造)
固化剂:己二酸二酰肼(大冢化学公司制造)
填充剂:二氧化硅球状微粒,SEAHOSTAR KE-C50HG(日本催化剂公司制造)
偶联剂:3-环氧丙氧基丙基三甲氧基硅烷,KBM-403(信越化学工业公司制造)
根据如表2和表4所示的结果可以确认:本发明的环氧树脂和部分酯化环氧树脂在液晶中的溶解性大幅降低。另外,根据如表4和图1~图6所示的取向性试验的结果,表明使用了本发明的环氧树脂的部分酯化环氧树脂即使在UV照射量为低照射量的情况下也不产生作为显示不良的原因的液晶取向紊乱,可知作为滴下工艺用液晶密封剂的低聚物成分而言有用性高。另一方面,在使用比较例5的组合物的情况下,在50mJ的照度下在拐角部因固化性组合物的溶出而产约200μm的液晶取向紊乱,此外在0mJ的照度下在拐角部和直线部均因固化性组合物的溶出而产生500~800μm的液晶取向紊乱。
产业上的可利用性
本发明的环氧树脂和部分酯化环氧树脂由于对液晶的溶解性、溶出性低,所以可用作通过紫外线等活性能量射线和热这两者而均可维持高可靠性的密封剂的原料。
Claims (7)
1.一种环氧树脂,其是由通式(1)、通式(2)或通式(3)表示的环氧树脂,

通式(1)、通式(2)和通式(3)中,
X为-O-、碳原子数1~4的烷撑基或碳原子数2~4的烷叉基,
Y为碳原子数1~4的烷撑基-碳原子数6~20的亚芳基-碳原子数1~4的烷撑基、碳原子数1~4的烷撑基-碳原子数6~20的亚芳基、或由-R7-(O-R7)n-表示的基团,所述由-R7-(O-R7)n-表示的基团中,R7为碳原子数1~4的烷撑基,n为0或1~6的整数,
R1、R2、R3、R4、R5和R6相互独立地为氢、缩水甘油基或甲基缩水甘油基,
各个R21各自相互独立地为氢或甲基,
R1、R2、R3、R4、R5和R6中的至少2个为缩水甘油基或甲基缩水甘油基。
2.一种部分酯化环氧树脂,其是由通式(4)、通式(5)或通式(6)表示的环氧树脂,

通式(4)、通式(5)和通式(6)中,
X、Y和R21如权利要求1中定义所述,
R11、R12、R13、R14、R15和R16为氢、缩水甘油基、甲基缩水甘油基、或由-Z-R8表示的基团,所述由-Z-R8表示的基团中,Z为2-羟基丙撑基或2-甲基-2-羟基丙撑基,R8为丙烯酰基或甲基丙烯酰基,
R11、R12、R13、R14、R15和R16中的至少2个为缩水甘油基、甲基缩水甘油基、或由-Z-R8表示的基团,
缩水甘油基和甲基缩水甘油基、与丙烯酰基和甲基丙烯酰基的比例,即,缩水甘油基+甲基缩水甘油基/丙烯酰基+甲基丙烯酰基,为10∶90~90∶10。
3.一种环氧树脂的制造方法,其包括工序(1A)~(1B),
工序(1A):使分子中具有2个以上的环氧基的多官能环氧化合物在金属催化剂的存在下与分子中具有2个以上的羟基的多羟基化合物发生反应,得到多官能环氧化合物的环氧开环体的工序,以及
工序(1B):使在工序(1A)中得到的多官能环氧化合物的环氧开环体的羟基发生环氧化的工序。
4.根据权利要求3所述的环氧树脂的制造方法,其是用于制造权利要求1所述的由通式(1)~通式(3)表示的环氧树脂的方法,包括下述工序(2A)~(2B),
工序(2A):使由通式(7a)、通式(8a)或通式(9a)表示的环氧化合物在金属催化剂的存在下与由下述通式(10)表示的二羟基化合物发生反应,从而得到由通式(7b)、通式(8b)或通式(9b)表示的环氧开环体的工序,


通式(7a)、通式(8a)和通式(9a)中,X和R21如权利要求1中定义所述,
HO-Y-OH (10)
通式(10)中,Y如权利要求1中定义所述,

通式(7b)、通式(8b)和通式(9b)中,X、Y和R21如权利要求1中定义所述;
工序(2B):使在工序(2A)中得到的由通式(7b)~通式(9b)表示的环氧开环体的羟基发生环氧化,得到由通式(1)~通式(3)表示的环氧树脂的工序。
5.一种部分酯化环氧树脂的制造方法,其包括工序(1C),
工序(1C):使通过权利要求3所述的制造方法制得的环氧树脂在碱性催化剂的存在下与(甲基)丙烯酸发生反应的工序。
6.根据权利要求5所述的部分酯化环氧树脂的制造方法,其是用于制造权利要求2所述的由通式(4)~通式(6)表示的部分酯化环氧树脂的方法,包括工序(2C),
工序(2C):使通过权利要求4所述的制造方法制得的由通式(1)、通式(2)、或通式(3)表示的环氧树脂在碱性催化剂的存在下与(甲基)丙烯酸发生反应,得到由通式(4)~通式(6)表示的部分酯化环氧树脂的工序,


通式(1)、通式(2)和通式(3)中,
X、Y、R1、R2、R3、R4、R5、R6、R21如权利要求1中定义所述,
R1、R2、R3、R4、R5和R6中的至少2个为缩水甘油基或甲基缩水甘油基。
7.一种固化性组合物,其含有选自(a)权利要求1所述的由通式(1)、通式(2)和通式(3)表示的环氧树脂,(b)权利要求2所述的由通式(4)、通式(5)和通式(6)表示的部分酯化环氧树脂,(c)通过权利要求3所述的制造方法制得的环氧树脂,以及(d)通过权利要求5所述的制造方法制得的部分酯化环氧树脂中的1种以上的树脂。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510587361.5A CN105131251B (zh) | 2010-10-01 | 2011-09-14 | 低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010223842A JP5736613B2 (ja) | 2010-10-01 | 2010-10-01 | 低溶出性エポキシ樹脂及びその部分エステル化エポキシ樹脂、その製造方法、並びにそれを含む硬化性樹脂組成物 |
| JP2010-223842 | 2010-10-01 | ||
| PCT/JP2011/070916 WO2012043222A1 (ja) | 2010-10-01 | 2011-09-14 | 低溶出性エポキシ樹脂及びその部分エステル化エポキシ樹脂、その製造方法、並びにそれを含む硬化性樹脂組成物 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201510587361.5A Division CN105131251B (zh) | 2010-10-01 | 2011-09-14 | 低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN103140535A true CN103140535A (zh) | 2013-06-05 |
| CN103140535B CN103140535B (zh) | 2015-09-02 |
Family
ID=45892693
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201510587361.5A Active CN105131251B (zh) | 2010-10-01 | 2011-09-14 | 低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 |
| CN201180047684.5A Active CN103140535B (zh) | 2010-10-01 | 2011-09-14 | 低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201510587361.5A Active CN105131251B (zh) | 2010-10-01 | 2011-09-14 | 低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP5736613B2 (zh) |
| KR (1) | KR101812832B1 (zh) |
| CN (2) | CN105131251B (zh) |
| TW (2) | TWI588173B (zh) |
| WO (1) | WO2012043222A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI724291B (zh) * | 2017-03-31 | 2021-04-11 | 日商協立化學產業股份有限公司 | 改質樹脂及含有其之硬化性樹脂組成物 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5979972B2 (ja) * | 2012-05-17 | 2016-08-31 | 協立化学産業株式会社 | エステル化エポキシ樹脂、その製造方法、及びそれを含む硬化性組成物 |
| JP6445780B2 (ja) * | 2013-05-09 | 2018-12-26 | 積水化学工業株式会社 | 液晶滴下工法用シール剤、上下導通材料、及び、液晶表示素子 |
| CN106796376A (zh) * | 2015-05-08 | 2017-05-31 | 积水化学工业株式会社 | 液晶显示元件用密封剂、上下导通材料及液晶显示元件 |
| WO2018056058A1 (ja) | 2016-09-26 | 2018-03-29 | 富士フイルム株式会社 | 投写型表示装置、投写表示方法、及び、投写表示プログラム |
| CN111560111A (zh) * | 2020-06-22 | 2020-08-21 | 陕西科技大学 | 一种bpa-ga酚醛环氧树脂及其制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002105147A (ja) * | 2000-09-28 | 2002-04-10 | Mitsui Chemicals Inc | 低臭気樹脂組成物よびそれを含む被覆材およびそれを用いた被覆工法 |
| JP2009185116A (ja) * | 2008-02-04 | 2009-08-20 | Air Water Inc | カルボキシル基含有エポキシアクリレート樹脂、それを含有するアルカリ現像可能な光硬化性・熱硬化性樹脂組成物およびその硬化物 |
| CN101747594A (zh) * | 2009-12-11 | 2010-06-23 | 上海新天和树脂有限公司 | 环氧丙烯酸预聚体树脂及其制法和在光固化涂料中的应用 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5936361B2 (ja) * | 1976-08-03 | 1984-09-03 | 三菱電機株式会社 | マイカ薄葉材 |
| DE2829236A1 (de) * | 1977-07-06 | 1979-01-25 | Ciba Geigy Ag | Lineare, glycidyl- und hydroxylgruppenhaltige polyaetherharze |
| JPH06157712A (ja) * | 1992-11-27 | 1994-06-07 | Nippon Kayaku Co Ltd | 変性エポキシ樹脂、エポキシ樹脂組成物及びその硬化物 |
| JP2001310927A (ja) * | 2000-04-28 | 2001-11-06 | Nippon Shokubai Co Ltd | 不飽和基含有カルボン酸化合物 |
| JP4524083B2 (ja) * | 2003-01-22 | 2010-08-11 | 阪本薬品工業株式会社 | 活性エネルギー線硬化性樹脂組成物 |
| JP4117838B2 (ja) * | 2003-02-14 | 2008-07-16 | 三井化学株式会社 | エポキシ部分エステル化物の製造方法 |
| JP4524127B2 (ja) * | 2004-03-18 | 2010-08-11 | 阪本薬品工業株式会社 | 新規なエポキシ樹脂およびそれを含む硬化性樹脂組成物 |
| JP4636593B2 (ja) * | 2004-08-27 | 2011-02-23 | 阪本薬品工業株式会社 | 熱硬化性エポキシ樹脂組成物 |
| JP4779362B2 (ja) * | 2005-01-06 | 2011-09-28 | 三菱化学株式会社 | 水素化エポキシ樹脂及びエポキシ樹脂組成物 |
| JP5172321B2 (ja) * | 2006-12-26 | 2013-03-27 | 三井化学株式会社 | 液晶シール剤 |
| JP5424021B2 (ja) * | 2009-03-04 | 2014-02-26 | Dic株式会社 | 繊維強化複合材料用樹脂組成物、その硬化物、プリント配線基板用樹脂組成物、繊維強化複合材料、繊維強化樹脂成形品、及びその製造方法 |
-
2010
- 2010-10-01 JP JP2010223842A patent/JP5736613B2/ja active Active
-
2011
- 2011-09-14 KR KR1020137011165A patent/KR101812832B1/ko active IP Right Grant
- 2011-09-14 CN CN201510587361.5A patent/CN105131251B/zh active Active
- 2011-09-14 CN CN201180047684.5A patent/CN103140535B/zh active Active
- 2011-09-14 WO PCT/JP2011/070916 patent/WO2012043222A1/ja active Application Filing
- 2011-09-27 TW TW105104930A patent/TWI588173B/zh active
- 2011-09-27 TW TW100134793A patent/TWI534170B/zh active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002105147A (ja) * | 2000-09-28 | 2002-04-10 | Mitsui Chemicals Inc | 低臭気樹脂組成物よびそれを含む被覆材およびそれを用いた被覆工法 |
| JP2009185116A (ja) * | 2008-02-04 | 2009-08-20 | Air Water Inc | カルボキシル基含有エポキシアクリレート樹脂、それを含有するアルカリ現像可能な光硬化性・熱硬化性樹脂組成物およびその硬化物 |
| CN101747594A (zh) * | 2009-12-11 | 2010-06-23 | 上海新天和树脂有限公司 | 环氧丙烯酸预聚体树脂及其制法和在光固化涂料中的应用 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI724291B (zh) * | 2017-03-31 | 2021-04-11 | 日商協立化學產業股份有限公司 | 改質樹脂及含有其之硬化性樹脂組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2012043222A1 (ja) | 2012-04-05 |
| TW201619231A (zh) | 2016-06-01 |
| KR20130118324A (ko) | 2013-10-29 |
| JP2012077202A (ja) | 2012-04-19 |
| JP5736613B2 (ja) | 2015-06-17 |
| CN105131251B (zh) | 2017-08-04 |
| TWI534170B (zh) | 2016-05-21 |
| TWI588173B (zh) | 2017-06-21 |
| CN105131251A (zh) | 2015-12-09 |
| KR101812832B1 (ko) | 2017-12-27 |
| CN103140535B (zh) | 2015-09-02 |
| TW201229081A (en) | 2012-07-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103140535B (zh) | 低溶出性环氧树脂及其部分酯化环氧树脂、它们的制造方法以及含有它们的固化性树脂组合物 | |
| CN104302686A (zh) | 酯化环氧树脂、其制造方法以及包含该酯化环氧树脂的固化性组合物 | |
| CN103140532B (zh) | 环氧树脂加合物及其热固性材料 | |
| CN102037049B (zh) | 环氧树脂的加合物及其制备方法 | |
| TWI662055B (zh) | 具有優良硬化後柔軟性的硬化性樹脂、(甲基)丙烯酸硬化性樹脂、及液晶密封劑組成物 | |
| CN116120519A (zh) | 一种超高纯度环氧树脂的精制方法 | |
| TWI586695B (zh) | Production method of propylene oxide ether compound, liquid crystal sealant and propylene oxide ether compound | |
| JP7194632B2 (ja) | 硬化性組成物およびその硬化物 | |
| JP2004182970A (ja) | 共重合ポリエーテル及びその製造法 | |
| CN102037045A (zh) | 衍生自非种子油基链烷醇酰胺的环氧树脂及其制备方法 | |
| JP2013234128A (ja) | エポキシ化合物及びその製造方法ならびにエポキシ樹脂組成物及びその硬化物 | |
| JP5897679B2 (ja) | 低溶出性エポキシ樹脂及びその部分エステル化エポキシ樹脂、その製造方法、並びにそれを含む硬化性樹脂組成物 | |
| KR20190064418A (ko) | 무수당 알코올 핵을 갖는 화합물 및 이의 제조 방법 | |
| JP7412939B2 (ja) | フェノキシ樹脂の製造方法 | |
| WO2022201985A1 (ja) | 変性エポキシ樹脂、その製造方法、硬化性樹脂組成物、その硬化物、塗料及び接着剤 | |
| JP2006111654A (ja) | 生分解性エポキシ樹脂組成物及びその硬化物 | |
| JP4857598B2 (ja) | エポキシ化合物、その製造方法及びエポキシ樹脂組成物 | |
| JP6910173B2 (ja) | ジチオカーボネート化合物およびこれを用いた樹脂組成物 | |
| JPH0940732A (ja) | 活性エネルギー線硬化型樹脂組成物 | |
| WO2023140256A1 (ja) | 変性エポキシ樹脂、硬化性樹脂組成物及びその硬化物 | |
| WO2024070871A1 (ja) | ビスフェノールの製造方法、エポキシ樹脂の製造方法、及びエポキシ樹脂硬化物の製造方法、並びにビスフェノール組成物 | |
| JP2024149214A (ja) | エポキシ樹脂及び再生繊維の製造方法、繊維含有エポキシ樹脂組成物の製造方法、並びに繊維含有エポキシ樹脂硬化物の製造方法 | |
| JPH0459815A (ja) | ポリマーポリオールの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |