CN101538228B - 抗流感和禽流感病毒药物化合物帕拉米韦的合成方法 - Google Patents
抗流感和禽流感病毒药物化合物帕拉米韦的合成方法 Download PDFInfo
- Publication number
- CN101538228B CN101538228B CN2008101018187A CN200810101818A CN101538228B CN 101538228 B CN101538228 B CN 101538228B CN 2008101018187 A CN2008101018187 A CN 2008101018187A CN 200810101818 A CN200810101818 A CN 200810101818A CN 101538228 B CN101538228 B CN 101538228B
- Authority
- CN
- China
- Prior art keywords
- amino
- alkene
- hydrochloric acid
- compound
- rwj
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(C)(C)*[C@@](C1)C=C[C@]1C(OC)=O Chemical compound CC(C)(C)*[C@@](C1)C=C[C@]1C(OC)=O 0.000 description 3
Images







Landscapes
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Description




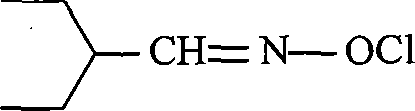
Claims (8)
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2008101018187A CN101538228B (zh) | 2008-03-21 | 2008-03-21 | 抗流感和禽流感病毒药物化合物帕拉米韦的合成方法 |
| HK10101112.9A HK1137416A1 (en) | 2008-03-21 | 2010-02-01 | Method for synthesizing medical compound peramivir for resisting influenza viruses and avian influenza viruses |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2008101018187A CN101538228B (zh) | 2008-03-21 | 2008-03-21 | 抗流感和禽流感病毒药物化合物帕拉米韦的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101538228A CN101538228A (zh) | 2009-09-23 |
| CN101538228B true CN101538228B (zh) | 2012-05-30 |
Family
ID=41121649
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2008101018187A Active CN101538228B (zh) | 2008-03-21 | 2008-03-21 | 抗流感和禽流感病毒药物化合物帕拉米韦的合成方法 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN101538228B (zh) |
| HK (1) | HK1137416A1 (zh) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102603577A (zh) * | 2011-01-19 | 2012-07-25 | 董慧珍 | 一种抗流感和禽流感药物的衍生物及其用途 |
| WO2012145932A1 (en) * | 2011-04-29 | 2012-11-01 | Pharmaresources (Shanghai) Co., Ltd. | A novel process for the preparation of peramivir and intermediates thereof |
| CN102757365B (zh) * | 2011-04-29 | 2013-10-30 | 上海泓博智源医药技术有限公司 | 一种制备帕拉米韦关键中间体的方法 |
| CN102863359B (zh) * | 2012-05-16 | 2014-05-07 | 常州制药厂有限公司 | 一种抗流感药物的合成方法 |
| CN102796056B (zh) * | 2012-08-23 | 2014-12-10 | 湖北丽益医药科技有限公司 | 帕拉米韦中间体及类似物的制备方法 |
| CN104072381B (zh) * | 2013-03-26 | 2017-04-12 | 安徽贝克联合制药有限公司 | 光学纯氨基醇盐酸盐的制备方法 |
| CN108997171B (zh) * | 2018-06-21 | 2020-11-27 | 苏州正济医药研究有限公司 | 一种3+2关环的合成方法 |
| CN114181117B (zh) * | 2020-09-15 | 2023-05-05 | 南京正济医药研究有限公司 | 一种帕拉米韦中间体的制备方法 |
| CN113880732A (zh) * | 2021-10-12 | 2022-01-04 | 湖南凯铂生物药业有限公司 | 帕拉米韦杂质a与杂质c及其制备方法和应用 |
| CN116425659B (zh) * | 2023-03-02 | 2023-11-03 | 浙江康聚药业有限公司 | 一种合成帕拉米韦的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1282316A (zh) * | 1997-12-17 | 2001-01-31 | 生物晶体药品股份有限公司 | 用作神经氨酸酶抑制剂的取代环戊烷和环戊烯化合物 |
| CN1358170A (zh) * | 1999-06-28 | 2002-07-10 | 生物晶体药品股份有限公司 | (一)-(1s,4r)n-保护4-氨基-2-环戊烯-1-羧酸酯的制备方法 |
-
2008
- 2008-03-21 CN CN2008101018187A patent/CN101538228B/zh active Active
-
2010
- 2010-02-01 HK HK10101112.9A patent/HK1137416A1/xx unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1282316A (zh) * | 1997-12-17 | 2001-01-31 | 生物晶体药品股份有限公司 | 用作神经氨酸酶抑制剂的取代环戊烷和环戊烯化合物 |
| CN1358170A (zh) * | 1999-06-28 | 2002-07-10 | 生物晶体药品股份有限公司 | (一)-(1s,4r)n-保护4-氨基-2-环戊烯-1-羧酸酯的制备方法 |
Non-Patent Citations (1)
| Title |
|---|
| 邱灵才等.抗流感及禽流感病毒新药"帕拉米韦"研究进展.《中国兽药杂志》.2006,第40卷(第6期),36-40. |
Also Published As
| Publication number | Publication date |
|---|---|
| HK1137416A1 (en) | 2010-07-30 |
| CN101538228A (zh) | 2009-09-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101538228B (zh) | 抗流感和禽流感病毒药物化合物帕拉米韦的合成方法 | |
| CN101967145B (zh) | 一种抗血栓药物阿匹沙班的制备方法 | |
| CN103524383B (zh) | 一种制备帕拉米韦的方法 | |
| CN103570580B (zh) | 一种高纯度碘普罗胺的制备方法 | |
| CN103664912B (zh) | 一种普卡必利的合成工艺 | |
| CN102746210A (zh) | 一种西洛多辛关键中间体的合成方法 | |
| CN112851646A (zh) | 特戈拉赞的制备方法 | |
| US9771317B2 (en) | Process for preparing lacosamide and related compounds | |
| CN110183357A (zh) | 一种用于制备沙库比曲中间体的制备方法 | |
| CN110655517A (zh) | 一种多替拉韦开环杂质的制备方法及其杂质 | |
| CN104311467B (zh) | 管式反应连续制备维格列汀的方法及装置 | |
| CN107698538A (zh) | 罗沙替丁醋酸酯盐酸盐的中间体3‑(1‑哌啶甲基)苯酚的新的制备方法 | |
| CN108864084B (zh) | 一组阿哌沙班有关物质及其制备方法 | |
| CN109810031A (zh) | 非罗考昔中间体的制备方法 | |
| CN113121412A (zh) | 一种雷芬那辛中间体的制备方法 | |
| CN107522691A (zh) | 一种chr20/21‑他克林杂合体化合物及其制备方法和应用 | |
| CN104761599B (zh) | 一种5,4’‑二羟基黄酮‑7‑o‑d‑葡萄糖醛酸的制备方法 | |
| CN101973909B (zh) | 一种米屈肼的制备方法 | |
| CN111116493B (zh) | 一种制备Apabetalone的方法、中间体及其中间体的制备方法 | |
| JP5092289B2 (ja) | 光学活性N−tert−ブチルカルバモイル−L−tert−ロイシンの製造方法 | |
| CN110577482B (zh) | 一种氨磺必利的制备方法 | |
| CN114702425A (zh) | (s)-2-氨基-(s)-3-[吡咯烷酮-2’]丙氨酸衍生物及中间体的制备方法 | |
| CN101654426B (zh) | 制备伊洛马司他的方法 | |
| JPS58172344A (ja) | フエニルアルカン酸の製造法 | |
| CN111533742A (zh) | 以氰胺为原料合成2-甲氧基三甲基嘌呤二酮的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1137416 Country of ref document: HK |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1137416 Country of ref document: HK |
|
| CP01 | Change in the name or title of a patent holder |
Address after: 100022, building 812, building A, zone 88, modern city, Jianguo Road, Beijing, Chaoyang District, Patentee after: BEIJING PUSHIKANG MEDICINE TECHNOLOGY Co.,Ltd. Patentee after: Tianjin Institute of Pharmaceutical Research Co.,Ltd. Address before: 100022, building 812, building A, zone 88, modern city, Jianguo Road, Beijing, Chaoyang District, Patentee before: BEIJING PUSHIKANG MEDICINE TECHNOLOGY Co.,Ltd. Patentee before: Tianjin Institute of Pharmaceutical Research |
|
| CP01 | Change in the name or title of a patent holder | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20210528 Address after: No.89, Shengsheng 1st Road, shuangd port, Dalian Economic and Technological Development Zone, Liaoning Province, 116651 Patentee after: DALIAN MAKING CHARM PHARMACEUTICAL Co.,Ltd. Patentee after: Tianjin Institute of Pharmaceutical Research Co.,Ltd. Address before: 100022, building 812, building A, zone 88, modern city, Jianguo Road, Beijing, Chaoyang District, Patentee before: BEIJING PUSHIKANG MEDICINE TECHNOLOGY Co.,Ltd. Patentee before: Tianjin Institute of Pharmaceutical Research Co.,Ltd. |
|
| TR01 | Transfer of patent right |