

Abstract
Although intermittent increases in inflammation are critical for survival during physical injury and infection, recent research has revealed that certain social, environmental and lifestyle factors can promote systemic chronic inflammation (SCI) that can, in turn, lead to several diseases that collectively represent the leading causes of disability and mortality worldwide, such as cardiovascular disease, cancer, diabetes mellitus, chronic kidney disease, non-alcoholic fatty liver disease and autoimmune and neurodegenerative disorders. In the present Perspective we describe the multi-level mechanisms underlying SCI and several risk factors that promote this health-damaging phenotype, including infections, physical inactivity, poor diet, environmental and industrial toxicants and psychological stress. Furthermore, we suggest potential strategies for advancing the early diagnosis, prevention and treatment of SCI.
One of the most important medical discoveries of the past two decades has been that the immune system and inflammatory processes are involved in not just a few select disorders, but a wide variety of mental and physical health problems that dominate present-day morbidity and mortality worldwide1–4. Indeed, chronic inflammatory diseases have been recognized as the most significant cause of death in the world today, with more than 50% of all deaths being attributable to inflammation-related diseases such as ischemic heart disease, stroke, cancer, diabetes mellitus, chronic kidney disease, non-alcoholic fatty liver disease (NAFLD) and autoimmune and neurodegenerative conditions5. Evidence is emerging that the risk of developing chronic inflammation can be traced back to early development, and its effects are now known to persist throughout the life span to affect adulthood health and risk of mortality6–8. In this Perspective, we describe these effects and out-line some promising avenues for future research and intervention.
Inflammation
Inflammation is an evolutionarily conserved process characterized by the activation of immune and non-immune cells that protect the host from bacteria, viruses, toxins and infections by eliminating pathogens and promoting tissue repair and recovery2,9. Depending on the degree and extent of the inflammatory response, including whether it is systemic or local, metabolic and neuroendocrine changes can occur to conserve metabolic energy and allocate more nutrients to the activated immune system9–12. Specific biobehavioral effects of inflammation thus include a constellation of energy-saving behaviors commonly known as “sickness behaviors,” such as sadness, anhedonia, fatigue, reduced libido and food intake, altered sleep and social-behavioral withdrawal, as well as increased blood pressure, insulin resistance and dyslipidemia10,13.These behavioral changes can be critical for survival during times of physical injury and microbial threat14.
A normal inflammatory response is characterized by the temporally restricted upregulation of inflammatory activity that occurs when a threat is present and that resolves once the threat has passed9,13,15. However, the presence of certain social, psychological, environmental and biological factors has been linked to the prevention of resolution of acute inflammation and, in turn, the promotion of a state of low-grade, non-infective (that is, ‘sterile’) systemic chronic inflammation (SCI) that is characterized by the activation of immune components that are often distinct from those engaged during an acute immune response13,16.
Shifts in the inflammatory response from short- to long-lived can cause a breakdown of immune tolerance9,15 and lead to major alterations in all tissues and organs, as well as normal cellular physiology, which can increase the risk for various non-communicable diseases in both young and older individuals1,9–11,15,17–21. SCI can also impair normal immune function, leading to increased susceptibility to infections and tumors and a poor response to vaccines22–25. Furthermore, SCI during pregnancy and childhood can have serious developmental consequences that include elevating the risk of non-communicable diseases over the life span7,8,26,27.
Systemic chronic inflammation and non-communicable disease risk
Although they share some common mechanisms, the acute inflammatory response differs from SCI (Table 1). Most notably, the acute inflammatory response is typically initiated during times of infection via an interaction between pattern recognition receptors expressed on innate immune cells and evolutionarily conserved structures on pathogens, called pathogen-associated molecular patterns (PAMPs). The acute inflammatory response can also be activated by damage-associated molecular patterns (DAMPs) that are released in response to physical, chemical or metabolic noxious stimuli—that is, ‘sterile’ agents—during cellular stress or damage2. Following infection, production of molecules such as lipoxins, resolvins, maresins and protectins then contribute to the resolution of inflammation28,29.
Table 1 |.
Acute inflammation versus systemic chronic inflammation
| Acute inflammation | Systemic chronic inflammation | |
|---|---|---|
| Trigger | PAMPs (infection), DAMPs (cellular stress, trauma) | DAMPs (‘exposome’, metabolic dysfunction, tissue damage) |
| Duration | Short-term | Persistent, non-resolving |
| Magnitude | High-grade | Low-grade |
| Outcome(s) | Healing, trigger removal, tissue repair | Collateral damage |
| Age-related | No | Yes |
| Biomarkers | IL-6, TNF-α, IL-1β, CRP | Silent—no canonical standard biomarkers |
DAMP, damage-associated molecular pattern; PAMP, pathogen-associated molecular pattern.
In contrast, SCI is typically triggered by DAMPs in the absence of an acute infectious insult or activation of PAMPs30–32. SCI often increases with age30, as indicated by studies showing that older individuals have higher circulating levels of cytokines, chemokines and acute phase proteins, as well as greater expression of genes involved in inflammation1,19,30. Moreover, SCI is low-grade and persistent (as the name suggests) and ultimately causes collateral damage to tissues and organs over time, such as by inducing oxidative stress1,4,9,19.
The clinical consequences of SCI-driven damage can be severe and include increased risk of the metabolic syndrome, which includes the triad of hypertension, hyperglycemia and dyslipidemia33,34; type 2 diabetes33; NAFLD33,35; hypertension1; cardiovascular disease (CVD)18,19; chronic kidney disease19; various types of cancer17; depression21; neurodegenerative and autoimmune diseases4,12,20; osteoporosis11,36 and sarcopenia19 (Fig. 1). Empirical evidence that inflammation plays a role in disease onset or progression is strongest for metabolic syndrome, type 2 diabetes and CVD. Indeed, it has long been known that patients with autoimmune diseases such as rheumatoid arthritis that are characterized by systemic inflammation have insulin resistance, dyslipidemia and hypertension, and that they have higher rates of metabolic syndrome, type 2 diabetes and CVD (particularly ischemic heart disease and str oke)10,12,37–39. Moreover, the inflammatory biomarker high-sensitivity C-reactive protein (CRP) is a predictor of cardiovascular events in men and women40. In a recent meta-analysis of data from more than 160,000 people across 54 long-term prospective studies, higher levels of circulating CRP were associated with a relative increase in risk for both coronary heart disease and CVD mortality41.
Fig. 1 |. Causes and consequences of low-grade systemic chronic inflammation.

Several causes of low-grade systemic chronic inflammation (SCI) and their consequences have been identified. As shown on the left, the most common triggers of SCI (in counter-clockwise direction) include chronic infections, physical inactivity, (visceral) obesity, intestinal dysbiosis, diet, social isolation, psychological stress, disturbed sleep and disrupted circadian rhythm, and exposure to xenobiotics such as air pollutants, hazardous waste products, industrial chemicals and tobacco smoking. As shown on the right, the consequences of SCI (in clockwise direction) include metabolic syndrome, type 2 diabetes, non-alcoholic fatty liver disease (NAFLD), cardiovascular disease, cancer, depression, autoimmune diseases, neurodegenerative diseases, sarcopenia, osteoporosis and immunosenescence.
The most compelling evidence for an association between SCI and disease risk comes from randomized controlled trials (RCTs) that have tested drugs or biologics that target specific pro-inflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α. In a recent meta-analysis of eight RCTs that included a total of 260 participants, anti-TNF-α inhibitor therapy was found to significantly reduce insulin resistance in patients with rheumatoid arthritis and to improve their insulin sensitivity42. The risk for developing Alzheimer’s disease was also significantly lower among patients with rheumatoid arthritis treated with the TNF-α inhibitor etanercept43. In addition, a recent double-blind RCT of the IL-1β inhibitor canakinumab that assessed more than 10,000 adults with a history of myocardial infarction and elevated circulating CRP levels showed that patients treated with canakinumab subcutaneously every 3 months had lower rates of nonfatal myocardial infarction, nonfatal stroke and CVD death compared with those treated with a placebo, despite having no change in LDL cholesterol, which is a risk factor for CVD. In this trial, canakinumab-treated patients also exhibited a lower likelihood of unstable angina leading to urgent revascularization44.
Along similar lines, a recent study of more than 160,000 people from North Glasgow found that a combination of the inflammatory markers CRP (>10 mg/L; HR 2.71, P < 0.001), albumin (>35 mg/L; HR 3.68, P < 0.001) and neutrophil count (HR 2.18, P < 0.001) predicted all-cause mortality over 8 years, in addition to mortality due to cancer, cardiovascular and cerebrovascular disease45.
Biomarkers for systemic chronic inflammation
Despite evidence linking SCI with disease risk and mortality45, there are presently no standard biomarkers for indicating the presence of health-damaging chronic inflammation. Studies have shown that canonical biomarkers of acute inflammation predict morbidity and mortality in both cross-sectional and longitudinal studies and may thus be used to index age-related SCI46. This approach has notable limitations, though. For example, early work by Roubenoff and colleagues showed that in monocytes from ambulatory individuals, levels of IL-6 and IL-1Ra (but not IL-1β or TNF-α) increased with age47. However, no difference in IL-1 and IL-6 expression has been found between young and older individuals when the health status of older individuals is strictly controlled48,49.
Additionally, a recent study examined levels of 18 endogenous and ex vivo-stimulated inflammatory markers in 41 healthy volunteers of different ages across the life span. Results revealed that unstimulated levels of IL-12p70 in women and CRP in men were associated with older age, whereas no effects were found for IL-1β, IFN-α or TNF-α50. Therefore, evidence exists that greater inflammatory activity is associated with older age, but this is not true of all inflammatory markers, and it is possible that these associations are due at least in part to increases in chronic ailments and frailty that are frequently associated with age rather than to biological aging itself.
To address limitations associated with assessing only a few select inflammatory biomarkers, some researchers have employed a multi-dimensional approach that involves assaying large numbers of inflammatory markers and then combining these markers into more robust indices representing heightened inflammatory activity. In one such study, researchers used principal component analysis to identify pro- and anti-inflammatory markers and an innate immune response that significantly predicted risk for multiple chronic diseases (CVD, kidney disease and diabetes), in addition to mortality51.
More recently, a multi-omics approach has been applied to examine links between SCI and disease risk. The researchers followed 135 adults longitudinally and conducted deep molecular profiling of participants’ whole-blood gene expression, termed the transcriptome; immune proteins—for example, cytokines and chemokines—termed the immunome; and cell subset frequencies, such as CD8+ T cell subsets, monocytes, natural killer (NK) cells, B cells and CD4+ T cell subsets. This enabled the researchers to construct a high-dimensional trajectory of immune aging (IMM-AGE) that described individuals’ immune functioning better than their chronological age. This new metric in turn accurately predicted all-cause mortality, establishing its potential future use for identifying at-risk patients in clinical settings52. These types of integrative, multi-level approaches to characterizing SCI are promising, but this work is still in its infancy and much more research is needed to identify best practices for selecting and analyzing SCI-related biomarkers in order to yield the most useful and predictive information for quantifying age-related disease risk.
Sources of systemic chronic inflammation
The SCI state in older individuals is thought to be caused in part by a complex process called cellular senescence, which is characterized by an arrest of cell proliferation and the development of a multifaceted senescence-associated secretory phenotype (SASP)53. A prominent feature of this phenotype is increased secretion of pro-inflammatory cytokines, chemokines and other pro-inflammatory molecules from cells53. Senescent cells expressing this phenotype can in turn promote a multitude of chronic health conditions and diseases, including insulin resistance, CVD, pulmonary arterial hypertension, chronic obstructive pulmonary disorder, emphysema, Alzheimer’s and Parkinson’s diseases, macular degeneration, osteoarthritis and cancer54,55.
How senescent cells acquire the SASP is not fully understood, but existing research points to a combination of both endogenous and non-endogenous social, environmental and lifestyle risk factors. Among the known endogenous causes of this phenotype are DNA damage, dysfunctional telomeres, epigenomic disruption, mitogenic signals and oxidative stress56. The non-endogenous contributors are thought to include chronic infections57, lifestyle-induced obesity58, microbiome dysbiosis59, diet60, social and cultural changes61,62 and environmental and industrial toxicants63. The fact that differences exist in the extent to which older adults exhibit SCI52,64 is thought to be indicative of inter-individual differences in exposure to these and other related pro-inflammatory factors, although studies documenting within-person associations between these risk factors and SCI are limited.
Nevertheless, differences in non-communicable diseases associated with SCI are evident across cultures and countries. Most prominently, SCI-related disease rates have increased dramatically for both older and younger individuals living in industrialized countries who follow a Western lifestyle but are relatively rare among individuals in non-Westernized populations who adhere to diets, lifestyles and ecological niches that more closely resemble those present during most of human evolution65–71. Furthermore, dietary and lifestyle habits, as well as exposure to a variety of different pollutants, can increase oxidative stress, upregulate mitogenic signaling pathways and cause genomic and epigenomic perturbations8,60,62,63 that can induce the SASP. Further evidence for a role of lifestyle in the development of chronic inflammation comes from a study of 210 healthy twins between 8 and 82 years old, which found that non-heritable factors are the strongest contributors to differences in chronic inflammation across individuals72 and that exposure to environmental factors, which have been collectively called the exposome, are the main drivers of SCI. Simply put, the exposome refers to a person’s lifelong exposure to physical, chemical and biological elements, starting from the prenatal period onward73.
Chronic infections.
The effect of lifelong infections caused by cytomegalovirus, Epstein–Barr virus, hepatitis C virus and other infectious agents on SCI and immune dysregulation remains controversial74–78. In terms of aging, chronic infection with cytomegalovirus has been associated with the so-called immune risk phenotype that has been predictive of early mortality in several longitudinal studies79. Furthermore, chronic infection with HIV causes premature aging of the immune system and is associated with early cardiovascular and skeletal changes57, with such effects being attributed in large part to the accumulation of senescent CD8+ T cells that produce increased levels of pro-inflammatory mediators80.
Although several studies have reported associations between chronic infections and autoimmune diseases, certain cancers, neurodegenerative diseases and CVD, chronic infections appear to interact synergistically with environmental and genetic factors to influence these health outcomes76,77,81. Indeed, humans coevolved with a variety of viruses, bacteria and other microbes82, and while chronic infections appear to contribute to SCI, they are not likely the primary driver. For instance, populations of hunter-gatherers and other existing non-industrialized societies such as the Shuar hunter-gatherers of the Ecuadorian Amazon83,84, Tsimané forager-horticulturalists of Bolivia68, Hadza hunter-gatherers from Tanzania67, subsistence agriculturalists from rural Ghana85 and traditional horticulturalists of Kitava (Papua New Guinea)86—all of whom are minimally exposed to industrialized environments but highly exposed to a variety of microbes—exhibit very low rates of inflammation-related chronic disease and substantial fluctuations in inflammatory markers that do not increase with age65,67,68,83,86.
Lifestyle, social and physical environment.
Individuals in the populations mentioned above have relatively short life expectancies on average, which means that some die before showing signs of advanced aging. However, the relative absence of SCI-related health problems in these populations has not been attributed to genetics or to having a shorter life expectancy, but rather to lifestyle factors and the social and physical environments the people inhabit66. Their lifestyles, for example, are characterized by higher levels of physical activity67,71,87, diets composed mainly of fresh or minimally processed food sources66,88,89, and less exposure to environmental pollutants66. In addition, individuals living in these environments generally have circadian rhythms that are more closely synchronized with diurnal fluctuations in sunlight exposure90 and the social stressors they experience are different from those typically present in industrialized environments91.
These social and environmental characteristics are believed to have predominated during most of hominin evolutionary history until industrialization66,82,89. Industrialization conferred many benefits, including social stability; reduced physical trauma; access to modern medical technology; and improved public health measures, such as sanitation, quarantine policies and vaccination, all of which significantly decrease infant mortality rates and increase average life expectancy66. However, more recently, these changes also caused radical shifts in diet and lifestyle, resulting in living circumstances that are very different from the ones that shaped human physiology for most of evolution. This is believed to have created an evolutionary mismatch in humans—characterized by an increasing separation from their ecological niche—and this mismatch, in turn, has been hypothesized to be a major cause of SCI65,66,82,89,92.
Physical activity.
Industrialization is thought to have caused a significant overall decrease in physical activity. One study showed that, worldwide, 31% of individuals are considered physically inactive—defined as not meeting the minimum international recommendations for regular physical activity—with levels of inactivity being higher in high-income countries than in low-to-middle-income countries93. In the United States, these numbers are even higher, with approximately 50% of American adults being considered physically inactive94.
Skeletal muscle is an endocrine organ that produces and releases cytokines and other small proteins, called myokines, into the bloodstream. This occurs particularly during muscle contraction and can have the effect of systemically reducing inflammation95. Low physical activity, therefore, has been found to be directly related to increased anabolic resistance96 and levels of CRP and pro-inflammatory cytokine levels in healthy individuals97, as well as in breast cancer survivors98 and patients with type 2 diabetes99. These effects can, in turn, promote several inflammation-related pathophysiologic alterations, including insulin resistance, dyslipidemia, endothelial dysfunction, high blood pressure and loss of muscle mass (sarcopenia)100, that have been found to increase risk for a variety of conditions, including CVD, type 2 diabetes, NAFLD, osteoporosis, various types of cancer, depression, dementia and Alzheimer’s disease, in individuals who are chronically inactive95,100.
Consistent with these effects, there is strong evidence for an association between physical inactivity and increased risk for age-related diseases and mortality. A recent meta-analysis of studies with cohorts from Europe, the United States and the rest of the world that included 1,683,693 participants found that going from physically inactive to achieving the recommended 150 minutes of moderate-intensity aerobic activity per week was associated with lower risk of CVD mortality by 23%, CVD incidence by 17%, and type 2 diabetes incidence by 26% during an average follow-up period of 12.8 years101. Moreover, data from 1.44 million participants across several prospective cohort studies revealed that, as compared to individuals exhibiting high levels of leisure-time physical activity (≥90th percentile), those who were physically inactive (≤10th percentile) had a greater risk (>20%) of developing several cancers, including esophageal adenocarcinoma; liver, lung, kidney, gastric cardia and endometrial cancers; and myeloid leukemia, even after adjusting for multiple major risk factors such as adiposity and smoking status (except for lung cancer)102. Likewise, a meta-analysis of ten studies and 23,345 older adults (70 to 80 years old) who were followed for 3.9–31 years found that individuals meeting the minimum international physical activity recommendations had a 40% lower risk of Alzheimer’s disease as compared to their physically inactive counterparts103.
Finally, physical inactivity can increase individuals’ risk for various non-communicable diseases because it is linked to obesity100 and, in particular, excessive visceral adipose tissue (VAT), which is a significant trigger of inflammation104–106. VAT is an active endocrine, immunological and metabolic organ composed of various cells (including immune cells, such as resident macrophages) that expands mostly through adipocyte hypertrophy, which can lead to areas of hypoxia and even cell death, resulting in activation of hypoxia-inducible factor-1α, increased production of reactive oxygen species, and release of DAMPs (for example, cell-free DNA). These events can induce the secretion of numerous pro-inflammatory molecules, including adipokines, cytokines (for example, IL-1β, IL-6, TNF-α), and chemokines (especially monocyte chemoattractant protein-1) by adipocytes, endothelial cells and resident adipose tissue immune cells (for example, macrophages)105–108. This in turn leads to the infiltration of various immune cells in the VAT, including monocytes, neutrophils, dendritic cells, B cells, T cells and NK lymphocytes, and a reduction in T regulatory cells, thereby amplifying inflammation, which can eventually become prolonged and systemic in some individuals106–109.
Furthermore, TNF-α and other molecules can cause adipocyte insulin resistance, which increases lipolysis, with the resulting spillover of lipids into other organs, such as the pancreas and liver, where they can contribute to beta-cell dysfunction, hepatic insulin resistance and fatty liver106. Hence, visceral obesity accelerates aging and increases risk for cardiometabolic, neurodegenerative and autoimmune diseases, as well as several types of cancer19,104,106,110–112. These dynamics are known to occur in adults and can promote age-related disease risk, but they first emerge during childhood26. The childhood obesity epidemic might thus be playing a key role in promoting inflammation and age-related disease risk worldwide113.
Microbiome dysbiosis.
Obesity may also lead to SCI through gut microbiome-mediated mechanisms114. For example, studies conducted in moderately obese Danish individuals without diabetes115 and in severely obese French women116 found changes in gut microbiota composition and microbial gene richness that were correlated with increased fat mass, pro-inflammatory biomarkers and insulin resistance. Furthermore, in older adults, changes in the gut microbiota seem to influence the outcome of multiple inflammatory pathways59.
Obesity, which is strongly linked to changes in the gut microbiome, has also been associated with increased intestinal paracellular permeability and endotoxemia114,117. Moreover, the latter is a suspected cause of inflammation through activation of pattern recognition receptors, such as Toll-like receptors, in immune cells and of inflammation-mediated metabolic conditions such as insulin resistance118. Interestingly, serum concentrations of zonulin, a protein that increases intestinal permeability, appear to be elevated in obese children and adults117,119, and in persons with type 2 diabetes118, NAFLD, coronary heart disease, polycystic ovary syndrome, autoimmune diseases and cancer117. More recently, elevated serum zonulin concentrations have been found to predict inflammation and physical frailty120.
More broadly, it has been hypothesized that a complex balance exists in the intestinal ecosystem that, if disrupted, can compromise its function and integrity and in turn cause low-grade SCI59. It may thus be important to identify possible triggers of dysbiosis and intestinal hyperpermeability, which could potentially include the overuse of antibiotics, nonsteroidal anti-inflammatory drugs and proton-pump inhibitors121,122; lack of microbial exposure induced by excessive hygiene and reduced contact with animals and natural soils, which is a very recent phenomenon in human evolutionary history82,123; and diet123 (see below).
Diet.
The typical diet that has become widely adopted in many countries over the past 40 years is relatively low in fruits, vegetables and other fiber- and prebiotic-rich foods66,123–125 and high in refined grains124, alcohol126 and ultra-processed foods125, particularly those containing emulsifiers127. These dietary factors can alter the gut microbiota composition and function123,127–130 and are linked to increased intestinal permeability129–131 and epigenetic changes in the immune system129 that ultimately cause low-grade endotoxemia and SCI129–131. The influence of diet on inflammation is not confined to these effects, though. For example, orally absorbed advanced glycation and lipoxidation end-products that are formed during the processing of foods or when foods are cooked at high temperatures and in low-humidity conditions are appetite increasing and are linked to overnutrition and hence obesity and inflammation132. Furthermore, high-glycemic-load foods, such as isolated sugars and refined grains, which are common ingredients in most ultra-processed foods, can cause increased oxidative stress that activates inflammatory genes133.
Other dietary components that are thought to influence inflammation include trans fatty acids134 and dietary salt. For example, salt has been shown to skew macrophages toward a pro-inflammatory phenotype characterized by the increased differentiation of naive CD4+ T cells into T helper (TH)-17 cells, which are highly inflammatory, and decreased expression and anti-inflammatory activity of T regulatory cells135. In addition, high salt intake can cause adverse changes in gut microbiota composition, as exemplified by the reduced Lactobacillus population observed in animals and humans fed high-salt diets135. This specific population is critical for health as it regulates TH17 cells and enhances the integrity of the intestinal epithelial barrier, thus reducing systemic inflammation135. Consistent with the expected health-damaging effects of consuming foods that are high in trans fats and salt, a recent cohort study of 44,551 French adults who were followed for a median of 7.1 years found that a 10% increase in the proportion of ultra-processed food consumption was associated with a 14% greater risk of all-cause mortality136.
Several other nutritional factors can also promote inflammation and potentially contribute to the development of SCI. These factors include deficiencies in micronutrients, including zinc137 and magnesium138, which are caused by eating processed or refined foods that are low in vitamins and minerals, and having suboptimal omega-3 levels139, which impacts the resolution phase of inflammation. Longchain omega-3 fatty acids—especially eicosapentaenoic acid and docosahexaenoic acid—modulate the expression of genes involved in metabolism and inflammation139. More importantly, they are precursors to molecules such as resolvins, maresins and protectins that are involved in the resolution of inflammation28,29. The main contributors to the growing worldwide incidence of low omega-3 status are a low intake of fish and high intake of vegetable oils that are high in linoleic acid, which displaces omega-3 fatty acids in cell membrane phospholipids140,141. In turn, various RCTs have shown that omega-3 fatty acid supplementation reduces inflammation142–144 and may thus have health-promoting effects141–144.
Evidence linking diet and mortality is robust. For example, an analysis of nationally representative health surveys and diseasespecific mortality statistics from the National Center for Health Statistics in the United States showed that the dietary risk factors associated with the greatest mortality among American adults in 2005 were high dietary trans fatty acids, low dietary omega-3 fatty acids, and high dietary salt145. In addition, a recent systematic analysis of dietary data from 195 different countries identified poor diet as the main risk factor for death in 2017, with excessive sodium intake being responsible for more than half of diet-related deaths146.
Finally, when combined with low physical activity, consuming hyperpalatable processed foods that are high in fat, sugar, salt and flavor additives147 can cause major changes in cell metabolism and lead to the increased production (and defective disposal) of dysfunctional organelles such as mitochondria, as well as to misplaced, misfolded and oxidized endogenous molecules30,60,148. These altered molecules, which increase with age19,30, can be recognized as DAMPs by innate immune cells, which in turn activate the inflammasome machinery, amplify the inflammatory response1,30,60 and contribute to a biological state that has been called “inflammaging,” defined as the “the long-term result of the chronic physiological stimulation of the innate immune system” that occurs in later life30. As proposed, inflammaging involves changes in numerous organ systems, such as the brain, gut, liver, kidney, adipose tissue and muscle19, and it is driven by a variety of molecular-age-related mechanisms that have been called the “Seven Pillars of Aging”55—namely, adaptation to stress, epigenetics, inflammation, macromolecular damage, metabolism, proteostasis and stem cells and regeneration.
Social and cultural changes.
In addition to physical inactivity and diet, the industrial revolution and modern era have ushered in changes in social interactions and sleep quality59,91 that can promote SCI149,150 and insulin resistance151, in turn increasing risk for obesity, type 2 diabetes, CVD and all-cause mortality150–154. Moreover, psychological stressors that are persistently present in some contemporary work environments, such as those characterized by high job demand and low control, can cause physiologic changes155 that disrupt the ability for glucocorticoids to effectively down-regulate inflammatory activity due to decreased sensitivity caused by chronic elevation in cortisol, leading in turn to SCI and poor health156.
Another core feature of modern society that has occurred very recently in human evolutionary history is increased exposure to artificial light, especially the blue spectrum, at atypical biologic times157–159. Exposure to blue light, especially after sundown, increases arousal and alertness at night and thus causes circadian rhythm disruption158,159, which in turn promotes inflammation160, and is a risk for multiple inflammation-related diseases157,159. As an example, night-shift work has been found to increase risk for the metabolic syndrome and is suspected of being a causal factor in obesity, type 2 diabetes and CVD, as well as in breast, ovarian, prostate, colorectal and pancreatic cancer157.
Environmental and industrial toxicants.
The rapid rise in urbanization over the past 200 years8 brought with it an unprecedented increase in humans’ exposure to various xenobiotics, including air pollutants, hazardous waste products and industrial chemicals that promote SCI8,161. Each year, an estimated 2,000 new chemicals are introduced into items that individuals use or ingest daily, including foods, personal care products, prescription drugs, household cleaners and lawn care products (see https://ntp.niehs.nih.gov). The concomitant increase in the estimated contribution of environmental chemicals to human disease burden162 has prompted a shift toward data generation using high-throughput screening to investigate the effect of industrial toxicants on cellular pathways, which has been supported by initiatives like the US Federal Tox21 Program, and toward the adoption of translational systems-toxicology approaches for integrating diverse data streams to better understand how chemicals affect human health and disease outcomes163. The Tox21 Program has tested more than 9,000 chemicals using more than 1,600 assays and has demonstrated that numerous chemicals to which people are commonly exposed greatly alter molecular signaling pathways that underlie inflammation and inflammation-related disease risk164. These chemicals include phthalates, per- and polyfluoroalkyl substances, bisphenols, polycyclic aromatic hydrocarbons and flame retardants165.
These compounds and others promote inflammatory activity via multiple mechanisms. For example, they can be cytotoxic8,162, cause oxidative stress or act as endocrine disruptors, starting in utero8. These chemicals are thus suspected of playing a causal role in hormone-dependent cancers, metabolic syndrome, type 2 diabetes, hypertension, CVD, allergy and asthma, and autoimmune and neurodegenerative diseases8,162,166. Tobacco smoking, which remains a worldwide health problem, is yet another source of xenobiotics that has been associated with a variety of inflammation-related diseases167.
Developmental origins of systemic chronic inflammation
The origins of SCI can also be viewed from a developmental perspective. For example, it is well established that childhood circumstances significantly impact metabolic and immune responses later in life, which in turn promote SCI in adulthood8,26,27,168,169. Childhood obesity, for instance, is strongly associated with major changes in adipose tissue and metabolic dysfunction that cause metabolism-related-SCI, or so-called metainflammation26. Because obese children often become obese adolescents and adults26, the risk of developing a pro-inflammatory phenotype also frequently persists into adulthood among individuals who were obese as children.
Another example of SCI being influenced by early life circumstances comes from epidemiologic studies showing that greater microbial exposure in infancy is associated with reduced risk of chronic inflammation in adulthood8,168, as predicted by the hygiene, or ‘old friends’, hypothesis82. Additionally, there is evidence that exposure to psychological stress early in life—for example, in the form of abuse, neglect, maltreatment, bullying or living in a low socioeconomic environment—can heighten neural responses to threat that can upregulate inflammatory activity170, alter immuno-competence and lead to SCI throughout the lifecycle27,169.
Further back in the developmental trajectory are data showing that the immune system is programmed during the prenatal period171 and is affected by epigenetic changes induced by maternal environmental exposures (for example, infectious agents, diet, psychological stress and xenobiotics) during intrauterine life and even before conception, when paternal factors may also have an epigenetic effect26,128,171. Together, these effects create the potential for the intergenerational transmission of risk for SCI. In this model (Fig. 2), SCI and disease risk are hypothesized to be perpetuated transgenerationally. In short, maternal inflammation during pregnancy172,173 is believed to pass an inflammatory ‘code’ through epigenetic modifications to the offspring, who will exhibit elevated risk for SCI in childhood and adulthood and therefore be more likely to suffer from a wide variety of inflammation-related health problems, including obesity7, CVD7, cancer174 and neurological illness175, among others, only to again pass this risk on to their own offspring.
Fig. 2 |. The maternal exposome and low-grade systemic chronic inflammation.
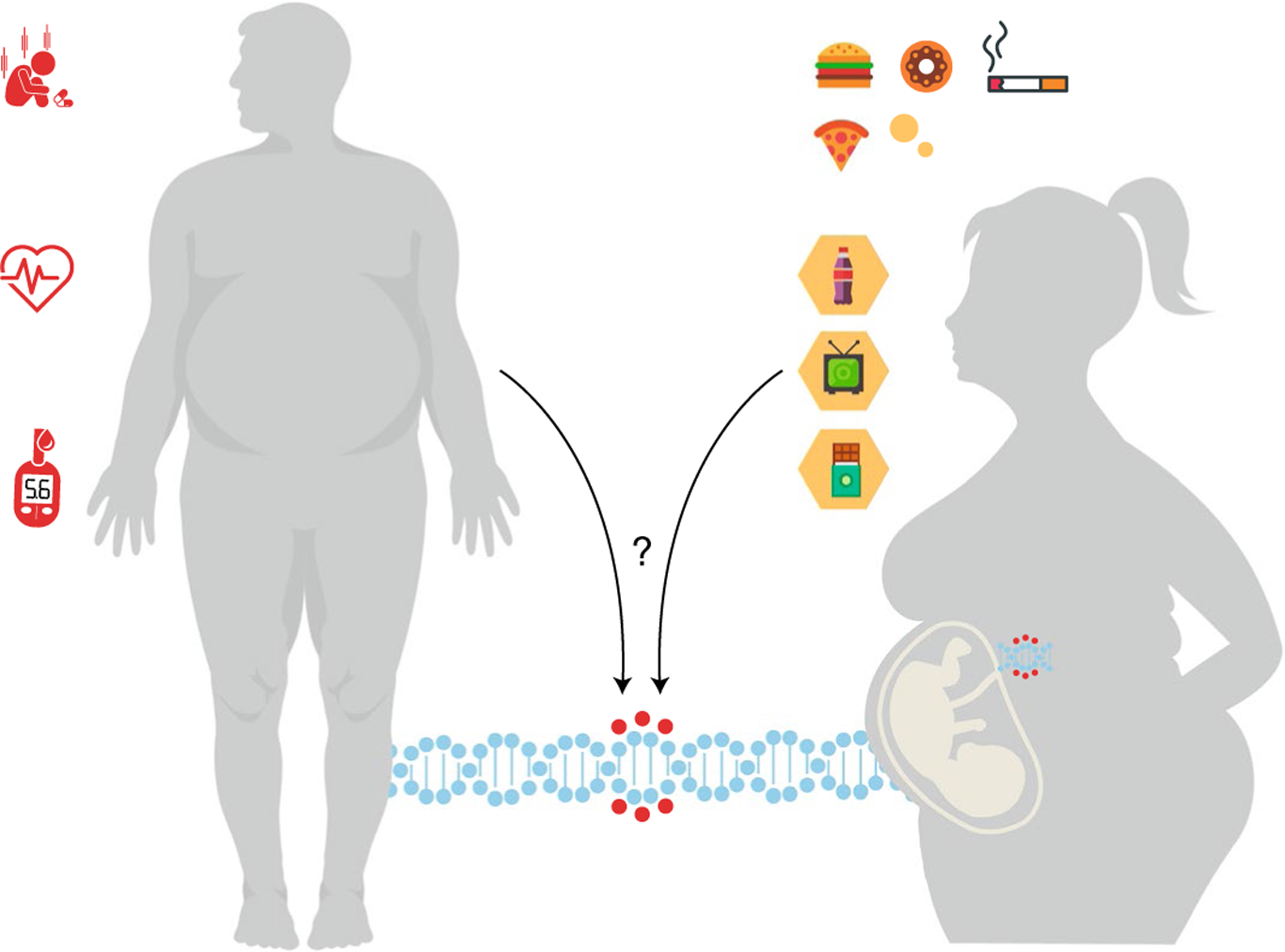
Maternal lifestyle and environmental exposures—collectively referred to as the exposome—include diet, physical activity, psychological stress and exposure to various xenobiotics, such as pollutants and smoking during intrauterine life. These factors in turn can influence the programming of the immune system of the offspring, potentially leading to a more pro-inflammatory phenotype later in life. Relevant factors, including environmental factors such as poor access to healthy food, housing insecurity, psychological stress and polluted air, lead to a mother giving birth to a fetus with epigenetic marks that increase the child’s risk for obesity, low-grade SCI and its associated consequences in adolescence and adulthood.
Chronic inflammation and the immune response to acute challenges
Despite the observation that SCI generally increases with age, a majority of older adults experience a down-regulation of components of the immune response that leads to an increased susceptibility to viral infections and weakened responses to vaccines. This apparent paradox (Fig. 3) can be explained by several mechanisms.
Fig. 3 |. Inflammatory model of immunosenescence and chronic disease.

This proposed model associates elevated baseline phosphorylated signaling proteins (for example, phosphorylated STAT (pSTAT) levels) with cellular unresponsiveness and chronically elevated inflammatory activity. The model involves an elevation of baseline pSTAT levels and its association with hallmark phenomenon of immunosenescence, an increased pro-inflammatory environment, unresponsive cells and a clinical impact on immune response. (Adapted with permission from ref.22, Elsevier.)
Specifically, elevated SCI can lead to a basal low-grade constitutive activation of various signaling pathways, such as the Janus kinase/signal transducers and activators of transcription (JAK–STAT) system in leukocytes, which results in a weakened acute response to multiple stimuli in immune cells from older adults with chronic inflammation due to reduced fold-increase in the levels of phosphorylation of these proteins after cell stimulation22. Elevated SCI has also been shown to predict hyporesponsiveness to the hepatitis B vaccine in humans24. Additionally, there is evidence that certain inflammatory biomarkers, such as CRP, are inversely correlated with older adults’ response to other vaccines, such as the herpes zoster vaccine23. Interestingly, this also seems to be true for younger individuals. Among adolescents, for example, those who respond well to typhoid vaccination have been found to exhibit lower concentrations of CRP than non-responders in adulthood25. In sum, this research helps to explain the pro-inflammatory/antiviral skewing that occurs as individuals age. This work also suggests that exposure to an inflammatory environment early in life is an important determinant of multiple aspects of an individual’s immuno-phenotype in adulthood.
Future directions
Considered together, this body of research provides converging evidence that SCI is associated with increased risk for developing a variety of chronic diseases that dominate present-day morbidity and mortality worldwide and that cause enormous amounts of human suffering. At the same time, there are several key avenues that could be pursued to help strengthen this work and translate this research into effective strategies for improving human health.
First, there is a clear need for additional studies that collect data on multiple factors affecting SCI to form a more comprehensive picture of how exposures and experiences identified at different levels of analysis combine to affect SCI and inflammation-related disease risk. Second, the field sorely needs robust integrative biomarkers of SCI that go beyond combining a few canonical biomarkers of acute inflammation. Existing biomarkers, which have primarily included CRP, IL-1β, IL-6 and TNF-α, have been useful for demonstrating that inflammatory activity is related to disease and mortality risk, but these markers provide only limited mechanistic information (given the enormous complexity of the inflammatory response) and they do not address anti-inflammatory regulatory pathways that may also be relevant for influencing inflammation-related disease risk. Future research should thus focus on additional biomarkers that have been found to have substantial variability across individuals, such as CD8+ T cell subsets, monocytes, NK cells, B cells and CD4+ T cell subsets176. This work should also include molecular, transcriptional and proteomic markers of SCI, which have only been examined in limited ways to date177. Constructing biomarkers that integrate information from a variety of different data sources and levels of analysis to represent inflammatory activity and immune regulation and dysregulation would be particularly useful, as would applying multi-omics approaches, computational modeling and artificial intelligence to study how SCI-related mechanisms both change and predict changes in clinical status within individuals over the life span178.
Third, given the difficulty associated with experimentally manipulating factors such as diet, sleep and stress levels that affect inflammation, a majority of studies conducted thus far have collected inflammatory biomarker data under basal conditions in which the immune system is not challenged. This is a sensible starting place, but such research does not provide any information regarding biological reactivity or recovery (for example, from infection or psychological or physiological stress), which may ultimately be most useful for understanding individual differences in inflammation-related disease risk3,179. Finally, although many of the SCI-promoting factors that we have described herein are at least partly modifiable—including physical inactivity, poor diet, night-time blue light exposure, tobacco smoking, environmental and industrial toxicants exposure and psychological stress—the number of studies that have successfully targeted these risk factors and shown corresponding reductions in SCI levels is limited. This has occurred despite the fact that the association between inflammation and chronic disease is now widely recognized and that healthcare systems are buckling due to the enormous cost of treating a worldwide population that is heavily burdened by SCI-related chronic health problems. Therefore, the time to start seriously studying how to prevent and treat SCI-related disease risk in both children and adults is now.
In conclusion, we have a long way to go before we fully understand the role that SCI plays in disease risk, biological aging and mortality. For example, no study to date has assessed the entire human exposome over the entire developmental trajectory, starting in utero (for example, by measuring maternal exposures, type of delivery and early-life nutrition) and continuing into adulthood (for example, by assessing social and cultural processes, xenobiotic exposures, individual lifestyle habits, lifelong antibiotic use, vaccinations, infectious diseases and social-psychological stressors). Moreover, only a few studies have investigated how modifying SCI-related processes may benefit human health or longevity. As a result, although SCI is a highly modifiable process in principle, additional research, initiative and investment are needed before we fully realize the potential benefits associated with targeting inflammation to improve human health.
Acknowledgements
This work was made possible by support from the National Institutes of Health (NIH) and the Buck Institute for Research on Aging to D.F., the National Institute on Aging, Glenn and SENS Foundations, and the Buck Institute for Research on Aging to J.C.; the Ministry of Education and Science of the Russian Federation Agreement (074-02-2018-330) and Horizon 2020 Framework Programme (634821, PROPAG-AGING) and JPco-fuND (ADAGE) to C.F.; the Intramural Research Program of the National Institute of Aging, NIH to L.F.; the MRC (UK) and Wellcome Trust to D.W.G.; NIH grant (R01 DK104344) to A.F.; the European Research Commission (PHII-669415), Associazione Italiana Ricerca sul Cancro (Projects IG 19014, 5×1000 9962 and 21147), Fondazione Cariplo, and Italian Ministry of Health to A.M.; NIH grant (P01 AG036695) to T.A.R.; the National Institute on Aging and UCLA AIDS Institute to R.B.E.; the Spanish Ministry of Economy and Competitiveness and Fondos FEDER (PI15/00558 and PI18/00139) to A.L.; and a Society in Science-Branco Weiss Fellowship, NARSAD Young Investigator Grant 23958 from the Brain & Behavior Research Foundation and NIH grant (K08 MH103443) to G.M.S. This work represents the opinion of the authors and does not reflect official NIH policy.
Footnotes
Peer review information Hannah Stower was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Furman D et al. Expression of specifc inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med 23, 174–184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Netea MG et al. A guiding map for inflammation. Nat. Immunol 18, 826–831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slavich GM Understanding inflammation, its regulation, and relevance for health: a top scientific and public priority. Brain Behav. Immun 45, 13–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett JM, Reeves G, Billman GE & Sturmberg JP Inflammation–nature’s way to efficiently respond to all types of challenges: implications for understanding and managing “the epidemic” of chronic diseases. Front. Med 5, 316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller GE, Chen E & Parker KJ Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychol. Bull 137, 959–997 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleming TP et al. Origins of lifetime health around the time of conception: causes and consequences. Lancet 391, 1842–1852 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renz H et al. An exposome perspective: early-life events and immune development in a changing world. J. Allergy Clin. Immunol 140, 24–40 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Kotas ME & Medzhitov R Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Straub RH, Cutolo M, Buttgereit F & Pongratz G Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J. Intern. Med 267, 543–560 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Straub RH, Cutolo M & Pacifici R Evolutionary medicine and bone loss in chronic inflammatory diseases—a theory of inflammation-related osteopenia. Semin. Arthritis Rheum 45, 220–228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Straub RH & Schradin C Chronic inflammatory systemic diseases: an evolutionary trade-of between acutely beneficial but chronically harmful programs. Evol. Med. Public Health 2016, 37–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Straub RH The brain and immune system prompt energy shortage in chronic inflammation and ageing. Nat. Rev. Rheumatol 13, 743–751 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Slavich GM Psychoneuroimmunology of stress and mental health in The Oxford Handbook of Stress and Mental Health (eds Harkness K & Hayden EP) (Oxford University Press, in the press; ). [Google Scholar]
- 15.Fullerton JN & Gilroy DW Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov 15, 551–567 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Calder PC et al. A consideration of biomarkers to be used for evaluation of inflammation in human nutritional studies. Br. J. Nutr 109, S1–S34 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Taniguchi K & Karin M NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol 18, 309–324 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Gisterå A & Hansson GK The immunology of atherosclerosis. Nat Rev. Nephrol 13, 368–380 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Ferrucci L & Fabbri E Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol 15, 505–522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heneka MT, Kummer MP & Latz E Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol 14, 463–477 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Miller AH & Raison CL The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol 16, 22–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen-Orr SS et al. Defective signaling in the JAK-STAT pathway tracks with chronic inflammation and cardiovascular risk in aging humans. Cell Syst. 3, 374–384.e4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verschoor CP et al. Serum C-reactive protein and congestive heart failure as signifcant predictors of herpes zoster vaccine response in elderly nursing home residents. J. Infect. Dis 216, 191–197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fourati S et al. Pre-vaccination inflammation and B-cell signalling predict age-related hyporesponse to hepatitis B vaccination. Nat. Commun 7, 10369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDade TW, Adair L, Feranil AB & Kuzawa C Positive antibody response to vaccination in adolescence predicts lower C-reactive protein concentration in young adulthood in the Philippines. Am. J. Hum. Biol 23, 313–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singer K & Lumeng CN The initiation of metabolic inflammation in childhood obesity. J. Clin. Invest 127, 65–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olvera Alvarez HA, Kubzansky LD, Campen MJ & Slavich GM Early life stress, air pollution, inflammation, and disease: an integrative review and immunologic model of social-environmental adversity and lifespan health. Neurosci. Biobehav. Rev 92, 226–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serhan CN Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serhan CN & Levy BD Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest 128, 2657–2669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franceschi C, Garagnani P, Vitale G, Capri M & Salvioli S Inflammaging and ‘garb-aging’. Trends Endocrinol. Metab 28, 199–212 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Liston A & Masters SL Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol 17, 208–214 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Frank D & Vince JE Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Difer. 26, 99–114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin C, Henao-Mejia J & Flavell RA Innate immune receptors: key regulators of metabolic disease progression. Cell Metab. 17, 873–882 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Hotamisligil GS Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Kazankov K et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol 16, 145–159 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Redlich K & Smolen JS Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov 11, 234–250 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Zhang J et al. The risk of metabolic syndrome in patients with rheumatoid arthritis: a meta-analysis of observational studies. PLoS One 8, e78151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armstrong AW, Harskamp CT & Armstrong EJ Psoriasis and the risk of diabetes mellitus: a systematic review and meta-analysis. JAMA Dermatol. 149, 84–91 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Dregan A, Charlton J, Chowienczyk P & Gulliford MC Chronic inflammatory disorders and risk of type 2 diabetes mellitus, coronary heart disease, and stroke: a population-based cohort study. Circulation 130, 837–844 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Ridker PM A test in context: high-sensitivity C-reactive protein. J. Am. Coll. Cardiol 67, 712–723 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Emerging Risk Factors Collaboration. et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet 375, 132–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burska AN, Sakthiswary R & Sattar N Effects of tumour necrosis factor antagonists on insulin sensitivity/resistance in rheumatoid arthritis: a systematic review and meta-analysis. PLoS One 10, e0128889 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou R et al. Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: a nested case-control analysis. CNS Drugs 30, 1111–1120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ridker PM et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Proctor MJ et al. Systemic inflammation predicts all-cause mortality: a Glasgow inflammation outcome study. PLoS One 10, e0116206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arai Y et al. Inflammation, but not telomere length, predicts successful ageing at extreme old age: a longitudinal study of semi-supercentenarians. EBioMedicine 2, 1549–1558 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roubenoff R et al. Monocyte cytokine production in an elderly population: effect of age and inflammation. J. Gerontol. A Biol. Sci. Med. Sci 53, M20–M26 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Ahluwalia N et al. Cytokine production by stimulated mononuclear cells did not change with aging in apparently healthy, well-nourished women. Mech. Ageing Dev 122, 1269–1279 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Beharka AA et al. Interleukin-6 production does not increase with age. J. Gerontol. A Biol. Sci. Med. Sci 56, B81–B8 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Elisia I et al. Effect of age on chronic inflammation and responsiveness to bacterial and viral challenges. PLoS One 12, e0188881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrisette-Tomas V et al. Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech. Ageing Dev 139, 49–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alpert A et al. A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat. Med 25, 487–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coppé J-P, Desprez P-Y, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu Y, Armstrong JL, Tchkonia T & Kirkland JL Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr. Opin. Clin. Nutr. Metab. Care 17, 324–328 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Kennedy BK et al. Geroscience: linking aging to chronic disease. Cell 159, 709–713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campisi J Aging, cellular senescence, and cancer. Annu. Rev. Physiol 75, 685–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Effros RB The silent war of CMV in aging and HIV infection. Mech. Ageing Dev 158, 46–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stout MB, Justice JN, Nicklas BJ & Kirkland JL Physiological aging: links among adipose tissue dysfunction, diabetes, and frailty. Physiology (Bethesda) 32, 9–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franceschi C, Garagnani P, Parini P, Giuliani C & Santoro A Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol 159, 1–15 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Zitvogel L, Pietrocola F & Kroemer G Nutrition, inflammation and cancer. Nat. Immunol 18, 843–850 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Razzoli M et al. Social stress shortens lifespan in mice. Aging Cell 17, e12778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carroll JE et al. Partial sleep deprivation activates the DNA damage response (DDR) and the senescence-associated secretory phenotype (SASP) in aged adult humans. Brain Behav. Immun 51, 223–229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan J et al. Long-term persistent organic pollutants exposure induced telomere dysfunction and senescence-associated secretary phenotype. J. Gerontol. A Biol. Sci. Med. Sci 73, 1027–1035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen-Orr SS & Furman D Variability in the immune system: of vaccine responses and immune states. Curr. Opin. Immunol 25, 542–547 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McDade TW Early environments and the ecology of inflammation. Proc. Natl Acad. Sci. USA 109, 17281–17288 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carrera-Bastos P, Fontes-Villalba M, O’Keefe JH, Lindeberg S & Cordain L The western diet and lifestyle and diseases of civilization. Res. Rep. Clin. Cardiol 2, 15–35 (2011). [Google Scholar]
- 67.Raichlen DA et al. Physical activity patterns and biomarkers of cardiovascular disease risk in hunter-gatherers. Am. J. Hum. Biol 29, e22919 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Kaplan H et al. Coronary atherosclerosis in indigenous South American Tsimane: a cross-sectional cohort study. Lancet 389, 1730–1739 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lindeberg S & Lundh B Apparent absence of stroke and ischaemic heart disease in a traditional Melanesian island: a clinical study in Kitava. J. Intern. Med 233, 269–275 (1993). [DOI] [PubMed] [Google Scholar]
- 70.Lindeberg S, Berntorp E, Nilsson-Ehle P, Terént A & Vessby B Age relations of cardiovascular risk factors in a traditional Melanesian society: the Kitava Study. Am. J. Clin. Nutr 66, 845–852 (1997). [DOI] [PubMed] [Google Scholar]
- 71.Lindeberg S, Eliasson M, Lindahl B & Ahrén B Low serum insulin in traditional Pacific Islanders—the Kitava Study. Metabolism 48, 1216–1219 (1999). [DOI] [PubMed] [Google Scholar]
- 72.Brodin P et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Niedzwiecki MM et al. The exposome: molecules to populations. Annu. Rev. Pharmacol. Toxicol 59, 107–127 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Virgin HW, Wherry EJ & Ahmed R Redefining chronic viral infection. Cell 138, 30–50 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Wang C et al. Effects of aging, cytomegalovirus infection, and EBV infection on human B cell repertoires. J. Immunol 192, 603–611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petta S et al. Hepatitis C virus infection is associated with increased cardiovascular mortality: a meta-analysis of observational studies. Gastroenterology 150, 145–155.e4 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Root-Bernstein R & Fairweather D Complexities in the relationship between infection and autoimmunity. Curr. Allergy Asthma Rep 14, 407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Furman D et al. Cytomegalovirus infection enhances the immune response to infuenza. Sci. Transl. Med 7, 281ra43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pawelec G et al. Human immunosenescence: is it infectious? Immunol. Rev 205, 257–268 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Chou JP, Ramirez CM, Wu JE & Efros RB Accelerated aging in HIV/AIDS: novel biomarkers of senescent human CD8+ T cells. PLoS One [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sochocka M, Zwolińska K & Leszek J The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol 15, 996–1009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rook G, Bäckhed F, Levin BR, McFall-Ngai MJ & McLean AR Evolution, human-microbe interactions, and life history plasticity. Lancet 390, 521–530 (2017). [DOI] [PubMed] [Google Scholar]
- 83.McDade TW et al. Analysis of variability of high sensitivity C-reactive protein in lowland Ecuador reveals no evidence of chronic low-grade inflammation. Am. J. Hum. Biol 24, 675–681 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Liebert MA et al. Implications of market integration for cardiovascular and metabolic health among an indigenous Amazonian Ecuadorian population. Ann. Hum. Biol 40, 228–242 (2013). [DOI] [PubMed] [Google Scholar]
- 85.Eriksson UK, van Bodegom D, May L, Boef AGC & Westendorp RGJ Low C-reactive protein levels in a traditional West-African population living in a malaria endemic area. PLoS One 8, e70076 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agmon-Levin N et al. Antitreponemal antibodies leading to autoantibody production and protection from atherosclerosis in Kitavans from Papua New Guinea. Ann. N. Y. Acad. Sci 1173, 675–682 (2009). [DOI] [PubMed] [Google Scholar]
- 87.Gurven M, Jaeggi AV, Kaplan H & Cummings D Physical activity and modernization among Bolivian Amerindians. PLoS One 8, e55679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cordain L et al. Plant-animal subsistence ratios and macronutrient energy estimations in worldwide hunter-gatherer diets. Am. J. Clin. Nutr 71, 682–692 (2000). [DOI] [PubMed] [Google Scholar]
- 89.Kuipers RS, Joordens JCA & Muskiet FAJ A multidisciplinary reconstruction of Palaeolithic nutrition that holds promise for the prevention and treatment of diseases of civilisation. Nutr. Res. Rev 25, 96–129 (2012). [DOI] [PubMed] [Google Scholar]
- 90.De la Iglesia HO et al. Ancestral sleep. Curr. Biol 26, R271–R272 (2016). [DOI] [PubMed] [Google Scholar]
- 91.Slavich GM & Cole SW The emerging field of human social genomics. Clin. Psychol. Sci 1, 331–348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chakravarthy MV & Booth FW Eating, exercise, and ‘thrifty’ genotypes: connecting the dots toward an evolutionary understanding of modern chronic diseases. J. Appl. Physiol 96, 3–10 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Hallal PC et al. Global physical activity levels: surveillance progress, pitfalls, and prospects. Lancet 380, 247–257 (2012). [DOI] [PubMed] [Google Scholar]
- 94.Katzmarzyk PT, Lee I-M, Martin CK & Blair SN Epidemiology of physical activity and exercise training in the United States. Prog. Cardiovasc. Dis 60, 3–10 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Fiuza-Luces C et al. Exercise benefits in cardiovascular disease: beyond attenuation of traditional risk factors. Nat. Rev. Cardiol 15, 731–743 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Breen L et al. Two weeks of reduced activity decreases leg lean mass and induces ‘anabolic resistance’ of myofibrillar protein synthesis in healthy elderly. J. Clin. Endocrinol. Metab 98, 2604–2612 (2013). [DOI] [PubMed] [Google Scholar]
- 97.Fedewa MV, Hathaway ED & Ward-Ritacco CL Effect of exercise training on C reactive protein: a systematic review and meta-analysis of randomised and non-randomised controlled trials. Br. J. Sports Med 51, 670–676 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Meneses-Echávez JF et al. The effect of exercise training on mediators of inflammation in breast cancer survivors: a systematic review with meta-analysis. Cancer Epidemiol. Biomarkers Prev 25, 1009–1017 (2016). [DOI] [PubMed] [Google Scholar]
- 99.Hayashino Y et al. Effects of exercise on C-reactive protein, inflammatory cytokine and adipokine in patients with type 2 diabetes: a meta-analysis of randomized controlled trials. Metab. Clin. Exp 63, 431–440 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Booth FW, Roberts CK & Laye MJ Lack of exercise is a major cause of chronic diseases. Compr. Physiol 2, 1143–1211 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wahid A et al. Quantifying the association between physical activity and cardiovascular disease and diabetes: a systematic review and meta-analysis. J. Am. Heart Assoc 5, e002495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moore SC et al. Association of leisure-time physical activity with risk of 26 types of cancer in 1.44 million adults. JAMA Intern. Med 176, 816–825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Santos-Lozano A et al. Physical activity and Alzheimer disease: a protective association. Mayo. Clin. Proc 91, 999–1020 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Pérez LM et al. ‘Adipaging’: ageing and obesity share biological hallmarks related to a dysfunctional adipose tissue. J. Physiol 594, 3187–3207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schipper HS, Prakken B, Kalkhoven E & Boes M Adipose tissueresident immune cells: key players in immunometabolism. Trends Endocrinol. Metabol 23, 407–415 (2012). [DOI] [PubMed] [Google Scholar]
- 106.Tchernof A & Després J-P Pathophysiology of human visceral obesity: an update. Physiol. Rev 93, 359–404 (2013). [DOI] [PubMed] [Google Scholar]
- 107.Pellegrinelli V, Carobbio S & Vidal-Puig A Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia 59, 1075–1088 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Frasca D, Blomberg BB & Paganelli R Aging, obesity, and inflammatory age-related diseases. Front. Immunol 8, 1745 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grant RW & Dixit VD Adipose tissue as an immunological organ. Obesity (Silver Spring) 23, 512–518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Versini M, Jeandel P-Y, Rosenthal E & Shoenfeld Y Obesity in autoimmune diseases: not a passive bystander. Autoimm. Rev 13, 981–1000 (2014). [DOI] [PubMed] [Google Scholar]
- 111.Himbert C et al. Signals from the adipose microenvironment and the obesity-cancer link–a systematic review. Cancer Prev. Res. (Phila.) 10, 494–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van Dijk G et al. Integrative neurobiology of metabolic diseases, neuroinflammation, and neurodegeneration. Front. Neurosci 9, 173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.NCD Risk Factor Collaboration (NCD-RisC). et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 390, 2627–2642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cani PD & Jordan BF Gut microbiota-mediated inflammation in obesity: a link with gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol 15, 671–682 (2018). [DOI] [PubMed] [Google Scholar]
- 115.Le Chatelier E et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013). [DOI] [PubMed] [Google Scholar]
- 116.Aron-Wisnewsky J et al. Major microbiota dysbiosis in severe obesity: fate afer bariatric surgery. Gut 68, 70–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sturgeon C & Fasano A Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 4, e1251384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jayashree B et al. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol. Cell. Biochem 388, 203–210 (2014). [DOI] [PubMed] [Google Scholar]
- 119.Küme T et al. The relationship between serum zonulin level and clinical and laboratory parameters of childhood obesity. J. Clin. Res. Pediatr. Endocrinol 9, 31–38 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qi Y et al. Intestinal permeability biomarker zonulin is elevated in healthy aging. J. Am. Med. Direc. Assoc 18, 810.e1–810.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Le Bastard Q et al. Systematic review: human gut dysbiosis induced by non-antibiotic prescription medications. Aliment. Pharmacol. Ter 47, 332–345 (2018). [DOI] [PubMed] [Google Scholar]
- 122.Bjarnason I et al. Mechanisms of damage to the gastrointestinal tract from nonsteroidal anti-inflammatory drugs. Gastroenterology 154, 500–514 (2018). [DOI] [PubMed] [Google Scholar]
- 123.Sonnenburg ED & Sonnenburg JL The ancestral and industrialized gut microbiota and implications for human health. Nat. Rev. Microbiol 17, 383–390 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Bentley J U.S. trends in food availability and a dietary assessment of loss-adjusted food availability, 1970–2014. EIB-166, U.S. Department of Agriculture, Economic Research Service (2017). [Google Scholar]
- 125.Martínez Steele E et al. Ultra-processed foods and added sugars in the US diet: evidence from a nationally representative cross-sectional study. BMJ Open 6, e009892 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grant BF et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: results from the national epidemiologic survey on alcohol and related conditions. JAMA. Psychiatry 74, 911–923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chassaing B, Van de Wiele T, De Bodt J, Marzorati M & Gewirtz AT Dietary emulsifers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut 66, 1414–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zmora N, Bashiardes S, Levy M & Elinav E The role of the immune system in metabolic health and disease. Cell Metab. 25, 506–521 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Richards JL, Yap YA, McLeod KH, Mackay CR & Mariño E Dietary metabolites and the gut microbiota: an alternative approach to control inflammatory and autoimmune diseases. Clin. Trans. Immunol 5, e82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bishehsari F et al. Alcohol and gut-derived inflammation. Alcohol Res. 38, 163–171 (2017). [PMC free article] [PubMed] [Google Scholar]
- 131.Lerner A & Matthias T Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimm. Rev 14, 479–489 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Vlassara H & Striker GE AGE restriction in diabetes mellitus: a paradigm shift. Nat. Rev. Endocrinol 7, 526–539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dickinson S, Hancock DP, Petocz P, Ceriello A & Brand-Miller J High-glycemic index carbohydrate increases nuclear factor-kappaB activation in mononuclear cells of young, lean healthy subjects. Am. J. Clin. Nutr 87, 1188–1193 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Mozafarian D, Aro A & Willett WC Health effects of trans-fatty acids: experimental and observational evidence. Eur. J. Clin. Nutr 63, S5–S21 (2009). [DOI] [PubMed] [Google Scholar]
- 135.Muller DN, Wilck N, Haase S, Kleinewietfeld M & Linker RA Sodium in the microenvironment regulates immune responses and tissue homeostasis. Nat. Rev. Immunol 19, 243–254 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Schnabel L et al. Association between ultraprocessed food consumption and risk of mortality among middle-aged adultsin France. JAMA Intern. Med 179, 490–498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bonaventura P, Benedetti G, Albarède F & Miossec P Zinc and its role in immunity and inflammation. Autoimm. Rev 14, 277–285 (2015). [DOI] [PubMed] [Google Scholar]
- 138.Nielsen FH Effects of magnesium depletion on inflammation in chronic disease. Curr. Opin. Clin. Nutr. Metab. Care 17, 525–530 (2014). [DOI] [PubMed] [Google Scholar]
- 139.Calder PC Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem. Soc. Trans 45, 1105–1115 (2017). [DOI] [PubMed] [Google Scholar]
- 140.Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF & Rawlings RR Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr 93, 950–962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Calder PC Very long-chain n-3 fatty acids and human health: fact, fiction and the future. Proc. Nutr. Soc 77, 52–72 (2018). [DOI] [PubMed] [Google Scholar]
- 142.Kiecolt-Glaser JK et al. Omega-3 supplementation lowers inflammation and anxiety in medical students: a randomized controlled trial. Brain Behav. Immun 25, 1725–1734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kiecolt-Glaser JK et al. Omega-3 supplementation lowers inflammation in healthy middle-aged and older adults: a randomized controlled trial. Brain Behav. Immun 26, 988–995 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.AbuMweis S, Jew S, Tayyem R & Agraib L Eicosapentaenoic acid and docosahexaenoic acid containing supplements modulate risk factors for cardiovascular disease: a meta-analysis of randomised placebo-control human clinical trials. J. Hum. Nutr. Diet 31, 67–84 (2017). [DOI] [PubMed] [Google Scholar]
- 145.Danaei G et al. The preventable causes of death in the United States: comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Med. 6, e1000058 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.GBD 2017 Diet Collaborators. Health effects of dietary risks in 195 countries, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 393, 1958–1972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hall KD Did the food environment cause theobesity epidemic? Obesity (Silver Spring) 26, 11–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.van Niekerk G, Toit du A, Loos B & Engelbrecht A-M Nutrient excess and autophagic deficiency: explaining metabolic diseases in obesity. Metab. Clin. Exp 82, 14–21 (2018). [DOI] [PubMed] [Google Scholar]
- 149.Slavich GM & Irwin MR From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol. Bull 140, 774–815 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tobaldini E et al. Short sleep duration and cardiometabolic risk: from pathophysiology to clinical evidence. Nat. Rev. Cardiol 16, 213–224 (2019). [DOI] [PubMed] [Google Scholar]
- 151.Reutrakul S & Van Cauter E Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metab. Clin. Exp 84, 56–66 (2018). [DOI] [PubMed] [Google Scholar]
- 152.Valtorta NK, Kanaan M, Gilbody S, Ronzi S & Hanratty B Loneliness and social isolation as risk factors for coronary heart disease and stroke: systematic review and meta-analysis of longitudinal observational studies. Heart 102, 1009–1016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Steptoe A, Shankar A, Demakakos P & Wardle J Social isolation, loneliness, and all-cause mortality in older men and women. Proc. Natl Acad. Sci. USA 110, 5797–5801 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kivimaki M & Steptoe A Efects of stress on the development and progression of cardiovascular disease. Nat. Rev. Cardiol 15, 215–229 (2018). [DOI] [PubMed] [Google Scholar]
- 155.Chandola T, Brunner E & Marmot M Chronic stress at work and the metabolic syndrome: prospective study. BMJ 332, 521–525 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cohen S et al. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc. Natl Acad. Sci. USA 109, 5995–5999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lunn RM et al. Health consequences of electric lighting practices in the modern world: a report on the National Toxicology Program’s workshop on shift work at night, artificial light at night, and circadian disruption. Sci. Total Environ 607–608, 1073–1084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hatori M et al. Global rise of potential health hazards caused by blue light-induced circadian disruption in modern aging societies. NPJ Aging Mech. Dis 3, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Touitou Y, Reinberg A & Touitou D Association between light at night, melatonin secretion, sleep deprivation, and the internal clock: health impacts and mechanisms of circadian disruption. Life Sci. 173, 94–106 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Leproult R, Holmbäck U & Van Cauter E Circadian misalignment augments markers of insulin resistance and inflammation, independently of sleep loss. Diabetes 63, 1860–1869 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jiang C et al. Dynamic human environmental exposome revealed by longitudinal personal monitoring. Cell 175, 277–291.e31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sly PD et al. Health consequences of environmental exposures: causal thinking in global environmental epidemiology. Ann. Glob. Health 82, 3–9 (2016). [DOI] [PubMed] [Google Scholar]
- 163.Collins FS, Gray GM & Bucher JR Toxicology. Transforming environmental health protection. Science 319, 906–907 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kleinstreuer NC et al. Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat. Biotechnol 32, 583–591 (2014). [DOI] [PubMed] [Google Scholar]
- 165.Tompson PA et al. Environmental immune disruptors, inflammation and cancer risk. Carcinogenesis 36, S232–S253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Floreani A, Leung PSC & Gershwin ME Environmental basis of autoimmunity. Clin. Rev. Allergy Immunol 50, 287–300 (2016). [DOI] [PubMed] [Google Scholar]
- 167.GBD 2015 Tobacco Collaborators. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990–2015: a systematic analysis from the Global Burden of Disease Study 2015. Lancet 389, 1885–1906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McDade TW, Rutherford J, Adair L & Kuzawa CW Early origins of inflammation: microbial exposures in infancy predict lower levels of C-reactive protein in adulthood. Proc. Biol. Sci 277, 1129–1137 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fagundes CP, Glaser R & Kiecolt-Glaser JK Stressful early life experiences and immune dysregulation across the lifespan. Brain Behav. Immun 27, 8–12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Slavich GM, Way BM, Eisenberger NI & Taylor SE Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc. Natl Acad. Sci. USA 107, 14817–14822 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Macpherson AJ, de Agüero MG & Ganal-Vonarburg SC How nutrition and the maternal microbiota shape the neonatal immune system. Nat. Rev. Immunol 17, 508–517 (2017). [DOI] [PubMed] [Google Scholar]
- 172.Blazkova J et al. Multicenter systems analysis of human blood reveals immature neutrophils in males and during pregnancy. J. Immunol 198, 2479–2488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Aghaeepour N et al. An immune clock of human pregnancy. Sci. Immunol 2, eaan2946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Simmen FA & Simmen RCM Te maternal womb: a novel target for cancer prevention in the era of the obesity pandemic? Eur. J. Cancer Prev 20, 539–548 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Le Belle JE et al. Maternal inflammation contributes to brain overgrowth and autism-associated behaviors through altered redox signaling in stem and progenitor cells. Stem Cell Reports 3, 725–734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Su LF et al. The promised land of human immunology. Cold Spring Harb. Symp. Quant. Biol 78, 203–213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Davis MM, Tato CM & Furman D Systems immunology: just getting started. Nat. Immunol 18, 725–732 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Schüssler-Fiorenza Rose SM et al. A longitudinal big data approach for precision health. Nat. Med 25, 792–804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Slavich GM & Sacher J Stress, sex hormones, inflammation, and major depressive disorder: extending social signal transduction theory of depression to account for sex differences in mood disorders. Psychopharmacology (Berl.) 236, 3063–3079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]