
Abstract
The krill species Euphausia superba plays a critical role in the food chain of the Antarctic ecosystem. Significant changes in climate conditions observed in the Antarctic Peninsula region in the last decades have already altered the distribution of krill and its reproductive dynamics. A deeper understanding of the adaptation capabilities of this species is urgently needed. The availability of a large body of RNA-seq assays allowed us to extend the current knowledge of the krill transcriptome. Our study covered the entire developmental process providing information of central relevance for ecological studies. Here we identified a series of genes involved in different steps of the krill moulting cycle, in the reproductive process and in sexual maturation in accordance with what was already described in previous works. Furthermore, the new transcriptome highlighted the presence of differentially expressed genes previously unknown, playing important roles in cuticle development as well as in energy storage during the krill life cycle. The discovery of new opsin sequences, specifically rhabdomeric opsins, one onychopsin, and one non-visual arthropsin, expands our knowledge of the krill opsin repertoire. We have collected all these results into the KrillDB2 database, a resource combining the latest annotation of the krill transcriptome with a series of analyses targeting genes relevant to krill physiology. KrillDB2 provides in a single resource a comprehensive catalog of krill genes; an atlas of their expression profiles over all RNA-seq datasets publicly available; a study of differential expression across multiple conditions. Finally, it provides initial indications about the expression of microRNA precursors, whose contribution to krill physiology has never been reported before.
Similar content being viewed by others


Introduction
Antarctic krill Euphausia superba represents a widely distributed crustacean of the Southern Ocean and one of the world’s most abundant species, with a total biomass between 100 and 500 million tonnes1. Due to its crucial ecological role in the Antarctic ecosystem, where it represents a link between apex predators and primary producers, several studies have been carried out over the years to characterize krill distribution2,3,4, population dynamics and structuring5, 6 and above all to understand its complex genetics5, 7,8,9. A sizable fraction of these studies focused on the DNA, specifically on mtDNA variation; however, the information available about krill genetics remains relatively modest. The difficulty in progressing this kind of study mainly depends on the considerable large krill genome size10, which is more than 15 times larger than the human genome. This aspect vastly complicates DNA sequencing, which is the reason why in recent years—together with the advances in high‐throughput RNA-sequencing techniques—different krill transcriptome resources have been developed11,12,13,14,15,16. However, it was with the KrillDB project17 that a detailed and advanced genetic resource was produced and made available to the community as an organized database. KrillDB is a web-based graphical interface with annotation results coming from the de novo reconstruction of the krill transcriptome, deriving from the assembly of more than 360 million Illumina sequence reads. In this study we significantly expanded the amount of input sequences, adding 45 new samples to those used in the previous work (see Table S3: Supplementary Material), for a total of more than 4 billion RNA-seq reads. We improved the transcriptome reconstruction strategy by merging multiple independent de novo assemblies into a unique reference through the use of filters and optimization procedures.
In addition, we updated KrillDB, now renamed KrillDB2 (available at https://krilldb2.bio.unipd.it/). We focused on two aspects: the improvement of the quality and breadth of the krill transcriptome sequences previously reconstructed, thanks to the addition of an unprecedented amount of RNA-sequencing data; and, correspondingly, an increase in the amount of information associated with each transcript. Each transcript annotation has been extended to include its splicing structure, the predictions of orthologs, its level of abundance in different sample groups, and finally the putative secondary structure in the case of microRNA precursors.
The new krill transcriptome increased our capability to identify transcriptional phenotypes previously undetected: for instance, we recovered a greater number of differentially expressed genes involved in cuticle development and in reproduction. The analyses performed also represent a crucial step forward in the characterization of E. superba opsins, providing a better snapshot of the complex mechanisms underlying the krill capability to adapt to extreme diel vertical migrations and seasonal changes in light availability.
Results
To create and annotate a de novo transcriptome assembly for Antarctic krill a preliminary investigation focusing on the efficiency and quality of already existing strategies for de novo transcriptome assembly of non-model organisms was performed. In a second step, we focused on identifying and applying the best transcriptome assembly strategy to finally explore the gene expression levels across different developmental stages and krill responses to different environmental conditions. At first, separate transcriptome reconstructions using different assembly programs were carried out. A combination of two filtering steps was applied to these results to discard artifacts and improve the assembly quality. Reconstructed transcripts across all assemblers were joined, producing a set of non-redundant representative transcripts. We obtained these results by applying the EvidentialGene pipeline (version 4), which was specifically designed to combine different reconstructions and to eliminate redundant sequences. Finally, we applied another filter to identify redundant or mis-assembled sequences still appearing in the transcriptome.
Transcriptome quality
We checked the quality of our reconstructed transcriptome step by step, starting from the independent de novo assemblies, then evaluating the potential of merging all assemblies into a unique meta-assembly, and finally filtering the transcriptome for redundancy. All these results are summarized in Fig. 1, Tables 1 and 2. The result of our reconstruction strategy was evaluated using different measures: the N50 statistics highlighted an increase in transfrag lengths at each step. Recent benchmarks, such as18, have shown that, while reconstructing the transcriptome of a species, no single approach is uniformly superior: the quality of each result is influenced by a number of factors, both technical (k-mer size, strategy for duplicate resolution) and biological (genome size, presence of contaminants). In our study, we observed that, although a consistent number of sequences was removed through each step of the assembly, merging and filtering procedure, we didn’t encounter any decline in the quality described by the basic statistics of the reconstructed transcripts (Table 1).

Transcriptome quality assessment results. Results of the first assembly filtering in terms of total number of transcripts.
We then explored the completeness of the krill transcriptome according to conserved ortholog content using BUSCO (version 4.0.5) comparing our sequences to all the expected single-copy orthologs from the Arthropoda phylum. The results of the BUSCO analyses performed on each independent de novo assembly, on the EvidentialGene reconstruction and the final transcriptome are reported in Table 2. This analysis confirms that our strategy for controlling redundancy did not affect transcriptome completeness: indeed, the fraction of complete single-copy essential genes dropped by 1.8% only, while 123,376 redundant transfrags were discarded.
We finally compared our quality assessment results with those from previously released krill transcriptomes (Table 3). Our latest assembly significantly improves all the metrics we have discussed above. While this evidence suggests that our assembly is reasonably close to providing a complete representation of the krill transcriptome, it is more difficult to gauge the amount of redundancy it contains. Specifically, it remains difficult to distinguish between splice variants of a gene and possible paralogous copies. We believe that only the availability of a genome draft will make it possible to reliably discriminate between these two signals.
Functional classification
The assembled fragments were aligned against known protein and nucleotide databases to understand whether they could be linked to specific functions or processes described in other species. The functional annotation analyses showed that 63,903 contigs (42% of the total krill transcriptome) matched at least one protein from the NCBI NR (non-redundant) collection for a total of 98,316 unique proteins, while 62,518 transfrags found homology with a UniProtKB/TREMBL protein sequences (41% of the total), matching a total of 96,005 unique proteins. Furthermore, 22,024 krill transcripts (15% of the total) had significant matches with sequences in the NCBI NT nucleotide database. To classify transcripts by putative function, we performed a GO assignment. Specifically, 2833 GO terms (corresponding to 13,064 genes) were assigned: 1224 of those (corresponding to 11,575 genes) represented molecular functions; 1193 terms (corresponding to 6990 genes) were linked to biological processes; 416 terms (corresponding to 4301 genes) represented cellular components.
A case study on the discovery of opsin genes
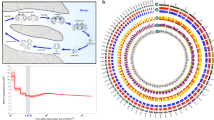
To evaluate the gene discovery potential of the new assembly, we searched the transcriptome for novel members of the opsin family. Opsins are a group of light sensitive G protein-coupled receptors with seven transmembrane domains. Fourteen genes were annotated as putative opsins, and the conserved domains analysis revealed that all of them possess the distinctive 7 α-helix transmembrane domain structure. The eight previously cloned opsins20 were all represented in KrillDB2 (sequence identity > 90%; Table S1 Supplementary Material). The other six genes we identified can therefore be considered new putative opsins. Among those, we found four putative rhabdomeric opsins: EsRh7 and EsRh8, with 70% and 59% of amino acid identity to EsRh1a and EsRh4, respectively; EsRh9 and EsRh10 showing high sequence identity (87% and 74%, respectively) to EsRh5. Furthermore, we identified two putative ancestral opsins: a non-visual arthropsin (EsArthropsin), and an onychopsin (EsOnychopsin) with 70% and 49% of sequence identity with crustacean and onychophoran orthologous, respectively. Phylogenetic analysis (Fig. 2) suggested that EsRh7-10 are middle-wavelength-sensitive (MWS) rhabdomeric opsins, and further confirmed EsArthropsin and EsOnychopsin annotation.

Phylogenetic relationships of Euphausia superba opsins shown as circular cladogram. Colored dots indicate krill opsins: red, previously cloned opsins; green, novel identified opsins. The spectral sensitivities of rhabdomeric opsin clades were inferred from the curated invertebrate-only opsin dataset proposed by DeLeo & Bracken‐Grissom, 2020. Represented opsin classes: LWS, long-wavelenght-sensitive; LSM, long/middle-wavelenght-sensitive; MWS, middle-wavelenght-sensitive; SWS/UV, short/UV-wavelenght-sensitive; ONY, onychopsins; MEL, melanopsins; PER, peropsin; ART, arthropsin. Rectangular phylogram is reported in Fig. S1 (Supplementary Material).
Differential expression
The availability of a new assembly of the krill transcriptome, reconstructed by collecting the largest amount of experimental data available thus far, suggested the possibility of performing a more detailed investigation of differential expression patterns. Therefore, we decided to reanalyze the dataset from Höring et al.21 to assess the possibility of identifying differentially expressed genes that were not detected in the original study due to the use of an older reference transcriptome15.
Our design matrix for the model included all the independent factors (season, area and sex) and, in addition, the interaction between area and season, sex and area, sex and season.
In total 1741 genes were differentially expressed (DEG) among experimental conditions. They correspond to around 2% of the total reconstructed genes. In the previous work by Höring21, the same samples were quantified against 58,581 contigs15 producing 1654 DEGs. Table 4 summarizes the list of performed contrasts, each one with the number of differentially expressed up and down regulated genes.
1195 DEGs were identified in the comparison between summer and winter specimens: 1078 were up-regulated and 117 down-regulated. In addition, 396 of such DEGs had some form of functional annotation. In general, these results are in accordance with the discussion by Höring21, which found that seasonal differences are predominant compared to regional ones. A summary of the DEGs is listed in Table 5. Complete tables of differentially expressed genes are downloadable on KrillDB2 (Fig. 3c; https://krilldb2.bio.unipd.it/, Section “Differentially Expressed Genes (DEGs)”).

Blast search section. The new search box for sequence searches (a) with an example of a BLAST search (highlighted in yellow) and the corresponding results (b). By clicking on each target identifier, the user will be redirected to that specific transcript page, where new sections have been added, as shown in Fig. 6.
Summer versus winter
We selected a series of genes among seasonal DEGs according to what has been already described in the literature. Höring et al.21 previously identified and described 35 relevant DEGs involved in seasonal physiology and behavior: we recovered the same gene signature in our analysis by comparing summer to winter samples. The majority of these DEGs appear to be involved in the development of cuticles (chitin synthase, carbohydrate sulfotransferase 11), lipid metabolism (fatty acid synthase 2, enoyl-CoA ligase), reproduction (vitellogenin, hematopoietic prostaglandin D synthase), metabolism of different hormones (type 1 iodothyronine deiodinase) and in the circadian clock (cryptochrome). Our results also include DEGs that were involved in the moult cycle of krill in other studies16. Specifically, we identified a larger group of genes involved in the different stages of the cuticle developmental process (peritrophin-A domain, calcified cuticle protein, glycosyltransferase 8-domain containing protein 1, collagen alpha 1, glutamine-fructose 6 phosphate), including proteins such as cuticle protein-3,6,19.8, early cuticle protein, pupal cuticle protein, endocuticle structural glycoprotein, chitinase-3 and chitinase-4, the latter representing a group of chitinase which have been shown to be expressed predominantly in gut tissue during larval and/or adult stages in other arthropods and are proposed to be involved in the digestion of chitin-containing substrates22. Finally, in addition to trypsin and crustin 4 (immune-related gene, essential in early pre-moult stage when krill still have a soft cuticle to protect them from pathogen attack, as seen by Seear et al.16), we also identified crustin-1,2,3,5 and 7. All the reported genes were up-regulated in summer, the period in which growth takes place and krill moult regularly.
Cuticle development genes were also identified as differentially expressed in the analysis of the interaction of multiple factors, between male samples coming from South Georgia and female specimens coming from the area of Bransfield Strait-South Orkney (considered as a unique area since they are placed at similar latitudes). Strikingly, we also identified a pro-resilin gene, whose role in many insects consists in providing efficient energy storage, being up-regulated in South Georgia male specimens.
Interaction effects
A number of relevant DEGs were found among specific regional and seasonal factors interactions. For instance, by comparing krill samples coming from South Georgia in summer and individuals sampled in Bransfield Strait-South Orkney in winter, we found genes up-regulated in summer in South Georgia related to reproductive activities, such as doublesex and mab-3 related transcription factor. The latter is a transcription factor crucial for sex determination and sexual differentiation, which was already described in other arthropods23. Since no differentially expressed gene related to reproduction was found by Höring et al.21 in the same comparisons, this suggests that the new krill transcriptome improves the power to identify new expression patterns and characterize the krill samples.
Finally, the comparison between male individuals from the Lazarev Sea and female specimens from the Bransfield Strait-South Orkney showed additional DEGs involved in reproduction, such as ovochymase 2, usually highly expressed in female adults or eggs, serine protease and a trypsin-like gene. In particular, trypsin-like genes are usually thought to be digestive serine proteases, but previous works suggested that they can play other roles24; many trypsins show female or male-specific expression patterns and have been found exclusively expressed in males, as in our analysis, suggesting that they play a role in the reproductive processes.
The simultaneous presence of differentially expressed genes involved in different steps of the krill moulting cycle, in the reproductive process and in sexual maturation that appear to be differentially expressed in the same comparisons is in accordance with what was already observed in krill25 and other krill species26. In particular, there is evidence of a strong relation between the krill moulting process and its growth and sexual maturation during the year, which supports and confirms the reliability of our results in terms of genes involved in such krill life cycle steps.
Identification of microRNA Precursors
Although microRNAs play a key role in the regulation of gene expression and in many important biological processes, such as development or cell differentiation, there is still no information about microRNAs in krill species.
Here we performed an investigation to test whether the new transcriptome could also include sequences with a significant homology to known mature microRNAs.
In total we identified 261 krill transcripts whose sequences are highly similar to 644 known microRNAs from other species. 306 sequences were linked to at least one GO term, matching 54 krill transcripts (Table S2, Supplementary Material). Among them, we identified 5 putative microRNAs involved with changes in cellular metabolism (age-dependent general metabolic decline—GO:0001321, GO:0001323), as well as changes in the state or activity of cells (age-dependent response to oxidative stress—GO:0001306, GO:0001322, GO:0001324), 35 microRNAs involved in interleukin activity and production. We found 26 putative microRNAs likely involved in ecdysteroidogenesis (specifically GO:0042768), a process resulting in the production of ecdysteroids, moulting and sex hormones found in many arthropods. In addition, we found a microRNA involved in fused antrum stage (GO:0048165) which appears to be related in other species to oogenesis. We also identified 27 microRNAs related to rhombomere morphogenesis, formation and development (GO:0021661, GO:0021663, GO:0021570). These functions have been linked to the development of portions of the central nervous system in vertebrates, which share the same structure of those found in arthropod brains. Lastly, 26 krill sequences showed high similarity with 2 mature microRNA related to the formation of tectum (GO:0043676), which represents in arthropods and, specifically, crustaceans, the part of the brain acting as visual center.
KrillDB2 web Interface
The KrillDB website has been redesigned to include the new version of the transcriptome assembly. Figures 3, 4, 5 and 6 collect images taken from the new main sections of the database. The integrated full-text search engine allows the user to search for a transcript ID, gene ID, GO term, a microRNA ID or any other free-form query. Results of full-text searches are now organized into several separate tables, each representing a different data source or biological aspect (Fig. 5). Results of GO term searches are summarized in a table reporting the related genes with corresponding domain or microRNA match and associated description. Both gene and transcript-centric pages have been extended with two new sections: “Orthology'' and “Expression” (Fig. 6). The Orthology section summarizes the list of orthologous sequences coming from the OMA analysis, each one with the species it belongs to and the identity score.

Differential Expression section. The new section collecting all differentially expressed genes tables (a) with an example of the corresponding result for a selected contrast (b).

New search engine of KrillDB2. Example of the results of a full-text search on KrillDB2.

Additional sections in gene and transcript pages. The new sections in the gene-centric page show a table listing the orthologous sequences with their belonging species and the identity score (a), a visualization of the gene structure as estimated by Lace software (d) and a boxplot coming from Expression Atlas analyses (c). Both Orthology and Expression sections are integrated also in the transcript-centric page. When a transcript is annotated as a putative microRNA, a “Predicted Hairpin” section displays a visualization of the hairpin predicted secondary structure and tables showing the alignment length, the HHMMiR score and the list of mature microRNAs matching (b).
The “Expression” section shows a barplot representing abundances estimates obtained from Salmon. An additional section, called “Gene Structure” (Fig. 6), was added to the gene page on the basis of the results coming from the SuperTranscript analysis. Specifically, we modified the STViewer.py Python script (from Lace), optimizing and adapting it to our own data and database structure, in order to produce a visualization of each gene with its transcripts. Since Lace relies on the construction of a single directed splice graph and it is not able to compute it for complex clusters with more than 30 splicing variants, this section is available for a selection of genes only.
The new KrillDB2 release includes completely updated transcript and gene identifiers. However, the user searching for a retired ID is automatically redirected to the page describing the newest definition of the appropriate transcript or gene.
The KrillDB2 homepage now includes two additional sections: one is represented by the possibility to perform a BLAST search (Fig. 3). Any nucleotide or protein sequence (query) can be aligned against krill sequences stored in the database. Results are summarized in a table containing information about the krill transcripts (target) that matched with the user’s query, and the e-value corresponding to the alignment. The other new section, called “Differentially Expressed Genes”, allows the user to browse all the tables listing the genes that were found to be differentially expressed among the conditions we have described above (Fig. 4). A drop-down menu gives access to the different comparisons; DEG tables list for each gene its log fold-change, p- and FDR values as estimated by edgeR. Moreover, each gene is linked to a functional description (if available) inferred from sequence homology searches.
Information about krill transcripts showing homology with an annotated microRNA is available in the “Predicted Hairpin” (Fig. 6). It contains a summary table with details about the hairpin length and the similarity score (as estimated by HHMMiR), followed by full listing of all the corresponding mature microRNAs (including links to their miRBase page). In addition, an image displaying the predicted secondary structure of the hairpin is included (computed by the “fornac” visualization software from the ViennaRNA suite).
Discussion
The availability of a large amount of public RNA-seq data capturing krill transcripts has allowed us to re-assemble its transcriptome and to significantly extend its annotation. We have now covered the entire developmental process of this species and included in our analysis individuals belonging to different seasons and affected by different environmental conditions. KrillDB2 provides the most complete source of information about the krill transcriptome and will offer a reliable starting point for the development of novel ecological studies. As shown in Fig. 1, Tables 1 and 2, the analysis of the quality of previously released krill transcriptome in comparison to the newly assembled KrillDB2 confirmed how the strategy applied did not produce any loss in terms of quality, although a consistent number of transcripts was removed. The quality metrics, in contrast, were improved both in terms of N50 statistics and transcriptome completeness: the fraction of complete single-copy essential genes reached 93.2%.
The differential expression analysis we have performed highlights the importance of specific processes in the complex krill life cycle and in its adaptation capability to the harsh Antarctic environment.
Identifying six novel putative opsin sequences almost doubles the eight previously cloned, demonstrating a significant improvement in the gene discovery potential of this new version of krill transcriptome. The finding of four novel MWS rhabdomeric opsins, an onychopsin, and a non-visual arthropsin further enrich the opsin repertoire of E. superba shedding light on a complex photoreception system able to coordinate the physiological and behavioral responses to the extreme daily (diel vertical migration) and seasonal changes in photoperiod and spectral composition. Arthropsins are rhabdomeric non-visual opsins and its clade is the sister group of the bilaterian rhabdomeric opsins27, 28. It was first discovered in the crustacean Daphnia pulex and subsequently in other arthropods, onychophoran, molluscs, annelids and flatworms27,28,29,30,31. Of relevance is the identification of an onychopsin which has been suggested to be the common ancestor of Panarthropoda visual opsins 27, and possibly sensitive to wavelength from UV to green light32. EsOnychopsin could represent the short-wavelength sensitive opsin (SWS/UV) which we have long been searching for. Indeed, the absence of a SWS/UV opsin was truly unexpected in an organism that shows daily vertical migration reaching depth beyond the 30 m, where only short wavelength light can penetrate.
Finally, KrillDB2 includes initial evidence about the presence of non-coding RNAs in krill, specifically sequences likely corresponding to microRNAs precursors. Although this is just a preliminary analysis, the results we have described already hint at a role of microRNAs in defining the adaptive capabilities of this species to the Antarctic environment. This represents a starting point for the study of non-coding RNAs in the Antarctic krill and in other species belonging to the same family.
Material and methods
Krill collection
This study aims at covering the entire developmental process of krill. Therefore, we used samples coming from different developmental stages to cover the entire E. superba transcriptome, from larval to adult specimens. Specifically, adults included both male and female specimens, as well as summer and winter individuals and they also came from 3 different geographical regions: Lazarev Sea, South Georgia, and Bransfield Strait/South Orkney. The entire samples collection used to produce the new transcriptomic reference and carry out all downstream analysis is listed in Table S3 (Supplementary Material).
Transcriptome assembly strategy
Multiple independent de novo assemblies
The assembly of short (Illumina) reads to reconstruct the transcriptomes of non-model organisms has been subject to a considerable amount of research. Out of the many tools developed for this task, we selected the five which are arguably the most popular in the field: Trinity (version 2.11.0)33, BinPacker (version 1.0)34, rnaSPAdes (version 3.14.1)35, TransABySS (version 2.0.1)36 and IDBA-tran (version 1.1.3)37. We summarized all the steps of the assembly reconstruction strategy, annotation process and downstream analyses in Fig. 7.

Workflow of the assembly process, annotation, database re-design and downstream analyses.
At first, we performed a separate transcriptome reconstruction with each of the tools listed above. Then, we evaluated their respective advantages through a series of independent measures, such as: the total number of transcripts; %GC content; the average fragment length; the total number of bases; the N50 value; and finally, the results of the BUSCO analysis, which provides a measure of transcriptome completeness based on evolutionarily informed expectations of gene content from near-universal single-copy orthologs38.
Assembly filtering and optimization
The raw sequencing data we used for the assemblies was obtained from different experiments and included both stranded (Table S3 Group 2) and unstranded libraries (Table S3 Group 1). As mixing these two types of libraries in a single assembly is not well supported, we decided to run each software twice: we thus generated a total of ten different de novo assemblies.
We used Trimmomatic39 to remove adapter sequences and other artifacts from raw Illumina sequences. The quality of trimmed reads was checked with the program FastQC40 (version 0.11.9). De novo transcriptome assembly was performed using specific parameters depending on the library type (the actual commands used are listed in Table S4, Supplementary Material).
Once assembled, a combination of two filtering steps was then applied to the newly reconstructed transcriptomes to discard artifacts and improve the assembly quality.
First, we estimated the abundances of all the transcripts reconstructed by each assembler using the Salmon software41 (version 1.4.0). Specifically, we used the following parameters were used: samples coming from unstranded library (Table S3, Group 1) were aligned using the options “-l ISR -1—validateMappings”; samples coming from stranded library (Table S3, Group 2) were aligned using the options “-l IU—validateMappings”. Samples were grouped according to the main experimental conditions: (1) sex, with female and male levels; (2) geographical area, covering Bransfield Strait, South Georgia, South Orkney and Lazarev Sea; and (3) season, with summer and winter levels. Abundance estimates were imported in the R statistical environment using the tximport package42 and we implemented a filter to keep only those transcripts showing an expression level of at least 1 transcript per million (TPM) within each of the three experimental conditions.
In a second step, we considered the results of all assemblers jointly, and we ran the “cd-hit-est” program (version 4.8.1)43, with parameters set as follows: -c 0.95-M 100000-T 22. This analysis was performed in order to cluster similar sequences and to produce a set of non-redundant representative transcripts. Specifically, we collapsed all sequences sharing 95% or more of their content, thus reducing the number of transcripts from 1,650,404 to 551,110.
Meta-assembly
The procedure described above was designed to identify near-duplicate sequences deriving from different software, but likely corresponding to the same biological transcript. As a further refinement, we were also interested in grouping resulting transcripts into units corresponding to genes. To this end, we relied on the EvidentialGene pipeline (version 4)44, 45. We applied the “tr2aacds” tool which clusters transcripts and classifies them to identify the most likely coding sequence representing each gene. EvidentialGene clustering was therefore applied using the following parameters: -NCPU = 22 -MAXMEM = 100000 -logfile -tidyup -species = Euphausia_superba. The software subdivides sequences into different categories, including primary transcript with alternates (main), primary without alternates (noclass), alternates with high and medium alignment to primary (althi1, althi, altmid) and partial (part) incomplete transcripts. A “coding potential” flag is also added, separating coding from non-coding sequences (see “KrillDB2 Web Interface” section). The meta-assembly thus obtained consisted in 274,840 putative transcripts, subdivided into 173,549 genes.
As these figures remained high, we performed another round of analyses to identify redundant or mis-assembled sequences still appearing in our transcriptome. Here we used a combination of BLAST searches against known protein and nucleotide databases (NR, NT, TREMBL) and information deriving from full-length, experimentally validated transcripts from a previous study46. Results confirmed that the newly reconstructed transcriptome fully represented krill RNAs, but the large amount of input reads, together with the number of independent de novo assemblers, likely led to an inflation in the number of alternative splicing variants being reconstructed. Moreover, transcript alignments against BUSCO genes38 and the doubletime, cry1, shaggy and vrille full-length transcripts from46 highlighted the fact that multiple fragments of the same gene were incorrectly assembled as separate transfrags. To remove these artifacts, first we aligned all transcript sequences in our meta-assembly against each other using the blastn tool. We discarded all sequences already included in a longer transcript for more than the 90% of their length. This filter helped us remove 78,731 redundant sequences (29% of transcripts, overall). Then, we ran a new abundance quantification using Salmon and we discarded all transcripts with an average abundance below 0.1 TPM.
The combination of all the filters discussed above allowed us to reduce the number of transcripts to 151,464 and, correspondingly, that of genes to 85,830. Our approach discarded redundant genes, while retaining alternative transcripts with a sufficient level of uniqueness in their sequence. This was confirmed by the fact that although we removed almost 45% of the initially assembled transcripts, this filtering barely affected the average read mapping rate, which went from 89% (initial EvidentialGene output) to 88% (full filtering). All samples appeared to be well represented in the reference transcriptome as confirmed by the fact that the average read mapping rates from each sample group was comparable (Group 1: 89%; Group 2: 88%; Group 3: 88%; Group 4: 90%).
In order to enhance the interpretability of the transcriptome reconstruction, we also employed a SuperTranscripts analysis, on the basis of the workflow proposed by47. Specifically, we ran the Lace software (https://github.com/Oshlack/Lace) to reconstruct the block structure of each gene (see “KrillDB2 Web Interface” section).
Functional annotation
Assembled fragments were aligned against the NCBI NR (non-redundant) and UniProtKB/TrEMBL protein databases, and against the NCBI NT nucleotide collection (data downloaded on 22/04/2021). We also ran InterproScan (version 5.51-85.0) to search for known functional domains and to predict protein family membership. Results with an e-value greater than 1e-6 for proteins (blastx) or 1e-9 for nucleotides (blastn) were discarded.
Gene orthology inference was performed using the Orthologus MAtrix (OMA) standalone package48 (https://omabrowser.org/standalone/) which relies on a complete catalog of orthologous genes among more than 2300 genomes covering the entire tree of life. This analysis helped us identify, based on protein sequences, those krill transcripts showing an orthology relationship with genes from other species and which sets of genes derived from a single common ancestral gene at a given taxonomic range49.
Finally, all krill transcripts were compared against the RNAcentral database (https://rnacentral.org/; https://doi.org/10.1093/nar/gkw1008) in order to identify any homology with the mature sequences of known microRNAs from other species.
Expression atlas
We used the final assembly described above to re-estimate transcript abundances over a wide range of RNAseq dataset (see Table S3) including:
-
Larval krill at two different stages of development exposed to different CO2 conditions, coming from17 (Table S3, Group 1)
-
Adult krill (48 samples) coming from different geographical areas (Bransfield Strait, Lazarev Sea, South Georgia, South Orkney) and different seasons (summer and winter), divided into male and female specimens21 (Table S3, Group 2)
-
Adult krill exposed to three different temperatures—Low Temperature, Mid temperature, High Temperature (Table S3, Group 3)
-
Adult krill divided into male and female specimens50 (Table S3, Group 4)
Overall, these datasets include six experimental factors: geographical area, season, developmental stage, pCO2 exposure condition, sex and temperature. Newly computed transcript abundances and raw counts were imported using R (version 4.0.5) and the package tximport (version 1.18.0). Batch effect removal was performed using the removeBatchEffect function implemented in the limma package (version 3.46.0). The resulting count matrix of transcripts (rows) across samples (columns) was then converted to the transcripts per million (TPM) scale. Finally, results were summarized to the gene level using the isoformToGeneExp function (IsoformSwitchAnalyzeR version 1.12.0). The expression levels for each experimental condition are displayed in KrillDB2 as a boxplot, as part of the webpage for each gene or transcript (see “KrillDB2 Web Interface” section).
Differential expression analysis
Transcript-level abundances and estimated counts were summarized at the gene-level using the package tximport. Resulting counts were normalized to remove unwanted variation by means of the RUVg method51. Specifically, we performed a preliminary between-sample normalization (EDASeq, version 2.24.0) to adjust for sequencing depth. Following the workflow outlined in the RUVseq vignette, we identified a set of negative control genes with an FDR level larger than 0.8. We applied the RUVg method to estimate k = 2 factors of unwanted variation and we included those in the design matrix for the final differential expression analysis, performed using the GLM method implemented by the edgeR software (version 3.32.1). All p-values were corrected using the Benjamini–Hochberg method.
MicroRNAs
We also investigated the possibility that the new transcriptome included sequences corresponding to the precursors of krill microRNAs.
To this aim, we ran the HHMMiR software52, which combines structural and sequence information to train a Hierarchical Hidden Markov Model for the identification of microRNA genes. We also performed a blastn search of all our assembled transcripts against the collection of miRBase (http://www.mirbase.org/) mature sequences. Results from these two analyses were combined: we collected all transcripts with a HHMMiR score below or equal to 0.71 and an alignment to a known mature microRNA with at most two mismatches. We then used the QuickGO tool (https://www.ebi.ac.uk/QuickGO/) to identify any potential association among our putatively identified microRNA precursors and GO categories.
Opsin phylogeny
To identify novel opsin genes in krill, we manually examined the list of transcripts that were annotated as “opsin” by our automated pipeline. Furthermore, the entire krill transcriptome was aligned against a curated opsin dataset (including 996 visual and non-visual opsins53) using Blast+(version 2.11.0). For genes with multiple alternative variants, we selected the longest transcript as a representative sequence. Secondary structure was assessed by the NCBI Conserved Domain Search (CDD database, May 2021). A phylogenetic tree was generated using the MUSCLE alignment tool and the Maximum Likelihood method (Dayhof substitution matrix and Nearest-Neighbor-Interchange method) as implemented in MEGA X (version 10.2.6, https://www.megasoftware.net/). New opsins were aligned against a curated invertebrate-only opsin data set54, the previously cloned krill opsins20, and the full-length onychopsin and arthropsin sequences available on the NCBI Protein database (May 2021, ncbi.nlm.nih.gov/protein). The tree was rooted using the human G protein-coupled receptor VIPR1 as an outgroup. Data S1 (Supplementary Material) includes the multi-alignments performed. Data S2 (Supplementary Material) contains all the protein sequences used to produce the tree.
Web interface implementation
The website was developed as a Python application based on the Flask framework. Data is stored in a PostgreSQL 12.8 database (http://www.postgresql.com). The sequences of the assembled transcripts and corresponding proteins are available for download as FASTA files. Gene and transcript pages have been updated with boxplots implemented using the Seaborn Python library (version 0.11.1).
Data availability
Data used for the krill transcriptome reconstruction and for the generation of the Expression Atlas was downloaded from the NCBI Short Read Archive, under accessions: PRJEB30084, PRJNA362526, PRJEB30084, PRJNA362526 and PRJNA640244.
Code availability
The scripts used in this research to assemble the krill transcriptome are listed in Table S4 (Supplementary Material).
References
Nicol, S., & Endo, Y. (1997). Krill fisheries of the world (No. 367). Food and Agriculture Org.
Atkinson, A. et al. Oceanic circumpolar habitats of Antarctic krill. Mar. Ecol. Prog. Ser. 362(1–23), 2008. https://doi.org/10.3354/meps07498 (2008).
Hofmann, E. E. & Murphy, E. J. Advection, krill, and Antarctic marine ecosystems. Antarct. Sci. https://doi.org/10.1017/s0954102004002275 (2004).
Siegel, V. Distribution and population dynamics of Euphausia superba: Summary of recent findings. Polar Biol. 29(1), 1–22. https://doi.org/10.1007/s00300-005-0058-5 (2005).
Bortolotto, E., Bucklin, A., Mezzavilla, M., Zane, L. & Patarnello, T. Gone with the currents: Lack of genetic differentiation at the circum-continental scale in the Antarctic krill Euphausia superba. BMC Genet. 12(1), 1–18. https://doi.org/10.1186/1471-2156-12-32 (2011).
Valentine, J. W. & Ayala, F. J. Genetic variability in krill. Proc. Natl. Acad. Sci. 73(2), 658–660. https://doi.org/10.1073/pnas.73.2.658 (1976).
Batta-Lona, P. G., Bucklin, A., Wiebe, P. H., Patarnello, T. & Copley, N. J. Population genetic variation of the Southern Ocean krill, Euphausia superba, in the Western Antarctic Peninsula region based on mitochondrial single nucleotide polymorphisms (SNPs). Deep Sea Res. Part II 58(13–16), 1652–1661. https://doi.org/10.1016/j.dsr2.2010.11.017 (2011).
Goodall-Copestake, W. P. et al. Swarms of diversity at the gene cox1 in Antarctic krill. Heredity 104(5), 513–518. https://doi.org/10.1038/hdy.2009.188 (2010).
Zane, L. et al. Molecular evidence for genetic subdivision of Antarctic krill (Euphausia superba Dana) populations. Proc. R. Soc. London Ser. B Biol. Sci. 265(1413), 2387–2391. https://doi.org/10.1098/rspb.1998.0588 (1998).
Jeffery, N. W. The first genome size estimates for six species of krill (Malacostraca, Euphausiidae): Large genomes at the north and south poles. Polar Biol. 35(6), 959–962. https://doi.org/10.1007/s00300-011-1137-4 (2012).
Clark, M. S. et al. Antarctic krill 454 pyrosequencing reveals chaperone and stress transcriptome. PLoS ONE 6(1), e15919. https://doi.org/10.1371/journal.pone.0015919 (2011).
De Pittà, C. et al. Systematic sequencing of mRNA from the Antarctic krill (Euphausia superba) and first tissue specific transcriptional signature. BMC Genom. 9(1), 1–14. https://doi.org/10.1186/1471-2164-9-45 (2008).
De Pittà, C. et al. The Antarctic krill Euphausia superba shows diurnal cycles of transcription under natural conditions. PLoS ONE 8(7), e68652. https://doi.org/10.1371/journal.pone.0068652 (2013).
Martins, M. J. F. et al. A transcriptome resource for Antarctic krill (Euphausia superba Dana) exposed to short-term stress. Mar. Genom. 23, 45–47. https://doi.org/10.1016/j.margen.2015.04.008 (2015).
Meyer, B. et al. Pyrosequencing and de novo assembly of Antarctic krill (Euphausia superba) transcriptome to study the adaptability of krill to climate-induced environmental changes. Mol. Ecol. Resour. 15(6), 1460–1471. https://doi.org/10.1111/1755-0998.12408 (2015).
Seear, P. J. et al. Differential gene expression during the moult cycle of Antarctic krill (Euphausia superba). BMC Genom. 11(1), 1–13. https://doi.org/10.1186/1471-2164-11-582 (2010).
Sales, G. et al. KrillDB: A de novo transcriptome database for the Antarctic krill (Euphausia superba). PLoS ONE 12(2), e0171908. https://doi.org/10.1371/journal.pone.0171908 (2017).
Hölzer, M. & Marz, M. De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-Seq assemblers. GigaScience 8(5), giz039. https://doi.org/10.1093/gigascience/giz039 (2019).
Hunt, B. J. et al. The Euphausia superba transcriptome database, Superba SE: An online, open resource for researchers. Ecol. Evol. 7(16), 6060–6077. https://doi.org/10.1002/ece3.3168 (2017).
Biscontin, A. et al. The opsin repertoire of the Antarctic krill Euphausia superba. Mar. Genomics 29, 61–68. https://doi.org/10.1016/j.margen.2016.04.010 (2016).
Höring, F. et al. Seasonal gene expression profiling of Antarctic krill in three different latitudinal regions. Mar. Genom. 56, 100806. https://doi.org/10.1016/j.margen.2020.100806 (2021).
Khajuria, C., Buschman, L. L., Chen, M. S., Muthukrishnan, S. & Zhu, K. Y. A gut-specific chitinase gene essential for regulation of chitin content of peritrophic matrix and growth of Ostrinia nubilalis larvae. Insect Biochem. Mol. Biol. 40(8), 621–629. https://doi.org/10.1016/j.ibmb.2010.06.003 (2010).
Jia, L. Y. et al. Doublesex evolution is correlated with social complexity in ants. Genome Biol. Evol. 10(12), 3230–3242. https://doi.org/10.1093/gbe/evy250 (2018).
Bao, Y. Y. et al. Genomic insights into the serine protease gene family and expression profile analysis in the planthopper, Nilaparvata lugens. BMC Genom. 15(1), 1–17. https://doi.org/10.1186/1471-2164-15-507 (2014).
Buchholz, F., Watkins, J. L., Priddle, J., Morris, D. J. & Ricketts, C. Moult in relation to some aspects of reproduction and growth in swarms of Antarctic krill, Euphausia superba. Marine Biol. 127(2), 201–208. https://doi.org/10.1007/BF00942104 (1996).
Tarling, G. A. & Cuzin-Roudy, J. Synchronization in the molting and spawning activity of northern krill (Meganyctiphanes norvegica) and its effect on recruitment. Limnol. Oceanogr. 48(5), 2020–2033. https://doi.org/10.4319/lo.2003.48.5.2020 (2003).
Hering, L. et al. Opsins in onychophora (velvet worms) suggest a single origin and subsequent diversification of visual pigments in arthropods. Mol. Biol. Evol. 29(11), 3451–3458. https://doi.org/10.1093/molbev/mss148 (2012).
Hering, L. & Mayer, G. Analysis of the opsin repertoire in the tardigrade Hypsibius dujardini provides insights into the evolution of opsin genes in Panarthropoda. Genome Biol. Evol. 6, 2380–2391. https://doi.org/10.1093/gbe/evu193 (2014).
Colbourne, J. K. et al. The ecoresponsive genome of Daphnia pulex. Science 331, 555–561. https://doi.org/10.1126/science.1197761 (2011).
Eriksson, B. J., Fredman, D., Steiner, G. & Schmid, A. Characterisation and localisation of the opsin protein repertoire in the brain and retinas of a spider and an onychophoran. BMC Evol. Biol. 13, 186. https://doi.org/10.1186/1471-2148-13-186 (2013).
Futahashi, R. et al. Extraordinary diversity of visual opsin genes in dragonflies. Proc. Natl. Acad. Sci. USA 112, E1247–E1256. https://doi.org/10.1073/pnas.1424670112 (2015).
Beckmann, H. et al. Spectral sensitivity in Onychophora (velvet worms) revealed by electroretinograms, phototactic behaviour and opsin gene expression. J. Exp. Biol. 218(6), 915–922. https://doi.org/10.1242/jeb.116780 (2015).
Grabherr, M. G. et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29(7), 644. https://doi.org/10.1038/nbt.1883 (2011).
Liu, J. et al. BinPacker: Packing-based de novo transcriptome assembly from RNA-seq data. PLoS Comput. Biol. 12(2), e1004772. https://doi.org/10.1371/journal.pcbi.1004772 (2016).
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8(9), giz100. https://doi.org/10.1093/gigascience/giz100 (2019).
Zhao, Q. Y. et al. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: A comparative study. BMC Bioinform. 12(14), 1–12. https://doi.org/10.1186/1471-2105-12-S14-S2 (2011).
Peng, Y. et al. IDBA-tran: A more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics 29(13), i326–i334. https://doi.org/10.1093/bioinformatics/btt219 (2013).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19), 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30(15), 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Andrews, S. (2017). FastQC: A quality control tool for high throughput sequence data. 2010.
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14(4), 417–419. https://doi.org/10.1038/nmeth.4197 (2017).
Love, M. I., Soneson, C. & Robinson, M. D. Importing transcript abundance datasets with tximport. Dim Txi. Inf. Rep. Sample 1(1), 5 (2017).
Li, W. & Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13), 1658–1659. https://doi.org/10.1093/bioinformatics/btl158 (2006).
Gilbert, D. G. Genes of the pig, Sus scrofa, reconstructed with EvidentialGene. PeerJ 7, e6374. https://doi.org/10.7717/peerj.6374 (2019).
Gilbert, D. G. (2019). Longest protein, longest transcript or most expression, for accurate gene reconstruction of transcriptomes? bioRxiv, 829184. https://doi.org/10.1101/829184
Biscontin, A. et al. Functional characterization of the circadian clock in the Antarctic krill Euphausia superba. Sci. Rep. 7(1), 1–13. https://doi.org/10.1038/s41598-017-18009-2 (2017).
Davidson, N. M., Hawkins, A. D. & Oshlack, A. SuperTranscripts: A data driven reference for analysis and visualization of transcriptomes. Genome Biol. 18(1), 1–10. https://doi.org/10.5281/zenodo.830594 (2017).
Altenhoff, A. M. et al. OMA standalone: Orthology inference among public and custom genomes and transcriptomes. Genome Res. 29(7), 1152–1163. https://doi.org/10.1101/gr.243212.118 (2019).
Altenhoff, A. M., Gil, M., Gonnet, G. H. & Dessimoz, C. Inferring hierarchical orthologous groups from orthologous gene pairs. PLoS ONE 8(1), e53786. https://doi.org/10.1371/journal.pone.0053786 (2013).
Suter, L. et al. Sex identification from distinctive gene expression patterns in Antarctic krill (Euphausia superba). Polar Biol. 42(12), 2205–2217. https://doi.org/10.1007/s00300-019-02592-3 (2019).
Risso, D., & Course, I. B. S. (2015). RNA-seq normalization and Batch effect removal.
Kadri, S., Hinman, V. & Benos, P. V. HHMMiR: Efficient de novo prediction of microRNAs using hierarchical hidden Markov models. BMC Bioinform. 10(1), 1–12. https://doi.org/10.1186/1471-2105-10-S1-S35 (2009).
Henze, M. J. & Oakley, T. H. The dynamic evolutionary history of pancrustacean eyes and opsins. Integr. Comp. Biol. 55(5), 830–842. https://doi.org/10.1093/icb/icv100 (2015).
DeLeo, D. M. & Bracken-Grissom, H. D. Illuminating the impact of diel vertical migration on visual gene expression in deep-sea shrimp. Mol. Ecol. 29(18), 3494–3510. https://doi.org/10.1111/mec.15570 (2020).
Acknowledgements
The position of Ilenia Urso was supported by the Helmholtz Virtual Institute "PolarTime": Biological timing in a changing marine environment—clocks and rhythms in polar pelagic organisms (VH-VI-500), headed by Bettina Meyer. Alberto Biscontin was funded by the “Programma Nazionale di Ricerche in Antartide – PNRA” (grant 2016_00225) and by the Promega Corporation 2019 Real-Time PCR Grant Program. We would also like to acknowledge the CAPRI initiative (Calcolo ad Alte Prestazioni per la Ricerca e l’Innovazione”, University of Padova Strategic Research Infrastructure Grant 2017) for the technical support and the HPC resources we have used for the analyses. Cristiano Bertolucci was supported by the “Programma Nazionale di Ricerche in Antartide – PNRA” (grant 2016_00225) and by the University of Ferrara research grant (FIR2020 and FAR2021).
Author information
Authors and Affiliations
Contributions
I.U., C.D.P., B.M. and G.S. conceived the study. I.U. and D.C. performed the analyses. I.U., A.B., B.M. and G.S. wrote the manuscript. C.B., C.R. and C.D.P. advised on data analysis and reviewed the text.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Urso, I., Biscontin, A., Corso, D. et al. A thorough annotation of the krill transcriptome offers new insights for the study of physiological processes. Sci Rep 12, 11415 (2022). https://doi.org/10.1038/s41598-022-15320-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15320-5
This article is cited by
-
Ecological genomics in the Northern krill uncovers loci for local adaptation across ocean basins
Nature Communications (2024)
-
Analysis of morphology, histology characteristics, and circadian clock gene expression of Onychostoma macrolepis at the overwintering period and the breeding period
Fish Physiology and Biochemistry (2024)
