
Abstract
Peptides exhibit lower affinity and a shorter half-life in the body than antibodies. Conversely, peptides demonstrate higher efficiency in tissue penetration and cell internalization than antibodies. Regardless of the pros and cons of peptides, they have been used as tumor-homing ligands for delivering carriers (such as nanoparticles, extracellular vesicles, and cells) and cargoes (such as cytotoxic peptides and radioisotopes) to tumors. Additionally, tumor-homing peptides have been conjugated with cargoes such as small-molecule or chemotherapeutic drugs via linkers to synthesize peptide–drug conjugates. In addition, peptides selectively bind to cell surface receptors and proteins, such as immune checkpoints, receptor kinases, and hormone receptors, subsequently blocking their biological activity or serving as hormone analogs. Furthermore, peptides internalized into cells bind to intracellular proteins and interfere with protein–protein interactions. Thus, peptides demonstrate great application potential as multifunctional players in cancer therapy.
Similar content being viewed by others


Introduction
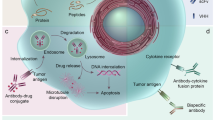
Compared with antibodies, peptides exhibit certain disadvantages, such as lower affinity, rapid excretion from the body (or shorter half-life in the body), and vulnerability to protease-mediated degradation. Conversely, the advantages of peptides include deep tissue penetration, efficient internalization into cells, lower immunogenicity and toxicity to the bone marrow and liver, and easy modification via chemicals compared with antibodies1,2,3,4. The pros and cons of peptides relative to antibodies are summarized in Table 1. Presently, >80 peptide therapeutics are available on the market; these include liraglutide (Victoza®), a glucagon-like peptide-1 used to treat type 2 diabetes mellitus, and leuprolide (Lupron®), a somatostatin analog used to treat prostate cancer4. In addition, peptides have been employed to identify peptide mimotopes5, generate vaccines6, and map protein–protein interaction epitopes7. Herein, we focused on the multifunctional application of peptides in targeted therapeutics; peptides can deliver carriers (such as nanoparticles, extracellular vesicles (EVs), and cells) and cargoes (such as cytotoxic peptides, radioisotopes, and small molecules) to target cells; inhibit or antagonize cell surface receptors and proteins; and interfere with intracellular protein–protein interactions. The various peptides, carriers, and cargoes described herein are summarized in Fig. 1.

Tumor-homing peptides bind to their receptors on tumor cells and selectively deliver cargoes therein, causing cell damage and death. Antagonist peptides target cell surface receptors on tumor cells, such as hormone receptor and PD-L1, and inhibit their biological activities. Interference peptides with or without tumor-homing peptides enter cells, bind to their intracellular targets, and inhibit the interaction between the target and its binding partner. Various types of carriers, such as nanoparticles, exosomes and cells, and cargoes, such as cytotoxic peptides, radionuclides, and drugs (PDCs), are used for targeted delivery via tumor-homing peptides.
Tumor-homing peptides as targeting ligands
Peptide-targeted delivery of nanoparticles
Tumor-homing peptides have been used for guiding nanoparticles to cancer cells through direct interactions between the peptides and receptors or binding partners on the cell surface8. In general, they are designed to be tumor cell-specific to enhance the internalization of nanoparticles into tumor cells. Multivalent labeling of peptides on nanoparticles increases the binding avidity of the peptide. In addition, the conjugation of peptides with nanoparticles tends to protect the peptide from protease-mediated degradation. The most well-known tumor-homing peptide is the RGD peptide, including RGD4C (ACDCRGDCFCG) and Cilengitide™ (RGDfV), which bind to overexpressed αvβ3 integrin in the angiogenic endothelial cells of tumor blood vessels, thereby inhibiting angiogenesis9,10,11. The conjugation of the RGD peptide with drugs or drug-loaded nanoparticles has been intensively investigated for cancer therapy12. Internalized RGD or iRGD (CRGDR/KGPDC), a modified version of the RGD peptide, not only bound to αV integrins but also increased the tissue penetration of drugs. The binding of the RGD motifs to the αV integrins expressed in tumor endothelial cells induces the protease-mediated cleavage of the iRGD peptide, producing two peptides, namely, CRGDR/K and GPDC. Subsequently, the CRGDR/K peptide containing the C-terminal CendR motif (R/KXXR/K) binds to neuropilin-1, activating an endocytic pathway13,14,15. Thus, iRGD increases the tissue penetration of drugs regardless of whether it is conjugated to or coadministered with the drug16,17,18.
The mitochondrial protein p32 or gC1qR is overexpressed in tumors with aberrant cell surface expression in tumor cells, tumor lymphatics, and a subset of myeloid cells such tumor-associated macrophages (TAMs)19. When conjugated with the p32-binding LyP-1 peptide (CGQKRTRGC), Abraxane, a nanoparticle albumin-bound paclitaxel, accumulated in tumor tissues and inhibited tumor growth more efficiently than untargeted Abraxane19,20. Vascular endothelial growth factor receptor 2 (VEGFR-2) is predominantly expressed on the surface of tumor endothelial cells21. Paclitaxel-loaded nanoparticles conjugated with K237 peptide (HTMYYHHYQHHL), a VEGFR-2-binding peptide, efficiently inhibited angiogenic activity and induced apoptosis of tumor endothelial cells and necrosis of tumor tissues22. Interleukin-4 receptor (IL4R), particularly type-II IL4R, is composed of IL4Rα and IL13Rα1, and it is upregulated in major tumors such as breast, lung, head and neck tumors and glioblastoma compared to their corresponding control tissues23,24,25. IL4RPep-1 peptide (CRKRLDRNC), an IL4R-binding peptide, can enhance the delivery of nanoparticles to IL4R-overexpressing tumors26,27,28,29,30. In addition, IL4R is highly expressed in M2-polarized, protumoral TAMs compared with M1-polarized, antitumoral macrophages, making IL4R a potential target for targeted drug delivery to TAMs31,32. The mannose receptor CD206 is also considered a cell surface marker of M2-type macrophages33. Nanoparticles labeled with a mUNO peptide (CSPGAK), which is a CD206-binding peptide, promote selective drug delivery to M2-type TAMs and induce M2 to M1 reprogramming of the macrophage phenotype33. The tumor-homing peptides used for the delivery of nanoparticles are summarized in Table 2.
Peptide-targeted delivery of EVs or exosomes
EVs or exosomes are endogenous nanoparticles secreted from cells into circulation. They can carry DNA, RNA, proteins, and lipids and distribute them among cells. Labeling tumor-homing peptides on the surface of exosomes loaded with therapeutics can reduce major adverse side effects in cancer therapy34. The surface modification of exosomes is performed via two methods: genetic and nongenetic engineering. Using genetic engineering, dendritic cells (DCs) have been engineered to secrete exosomes expressing Lamp2, an exosomal membrane protein, fused with the neuron-specific RVG peptide (YTIWMPENPRPGTPCDIFTNSRGKRASNG)35. Subsequently, RVG peptide-guided exosomes were employed to deliver short interfering RNA to neurons, microglia, and oligodendrocytes in the brain by targeting the gamma-aminobutyric acid (GABA) receptor, inducing target gene knockdown with negligible nonspecific uptake in other tissues35. Similarly, mouse immature DCs were genetically engineered to secrete exosomes expressing the Lamp2 protein fused with the iRGD peptide (CRGDR/KGPDC) and demonstrated highly efficient targeted drug delivery to αv integrin-positive breast cancer cells, consequently inhibiting tumor growth36. Cellular exosomes engineered to express the transmembrane domain of platelet-derived growth factor receptor fused with the GE11 peptide (YHWYGYTPQNVI), an epidermal growth factor receptor (EGFR)-binding peptide, selectively delivered let-7a microRNA to breast cancer tissues37. Moreover, tumor cell-derived exosomes genetically engineered to express a pH-sensitive fusogenic GALA peptide (WEAALAEALAEALAEHLAEALAEALEALAA) efficiently delivered tumor antigens to the cytoplasm of DCs and promoted the tumor antigen presentation of DCs via the major histocompatibility complex class I molecule38.
Exosomal surfaces have also been nongenetically modified using lipid-based membrane anchors, electrostatic interactions, and ligand–receptor interactions. M1 macrophage-derived exosomes were transfected with NF-κB p50 siRNA and miR-511-3p to foster M1 polarization and subjected to surface modification with the IL4R-targeting IL4RPep-1 peptide (CRKRLDRNC) using a phospholipid anchor; these constructs inhibited tumor progression by reprogramming IL4R-high and M2-polarized TAMs to an M1-like phenotype39. The surface modification of blood exosomes with transferrin-conjugated superparamagnetic nanoparticles via interaction with the transferrin receptor, with L17E endosomolytic peptides (IWLTALKFLGKHAAKHEAKQQLSKL) via electrostatic interactions, and with cholesterol-conjugated miR-21 inhibitor by anchoring to the lipid membrane increased tumor accumulation and drug delivery and enabled efficient endosomal escape40. Exosome surface labeling with a chimeric peptide (C16K-protoporphyrin IX-PKKKRKV) comprising an alkyl chain (C16), photosensitizer (protoporphyrin IX), and nuclear localization signal peptide (PKKKRKV) can enhance the nuclear delivery of the photosensitizer and efficiently inhibit tumor growth via photodynamic therapy41. The tumor-homing peptides used for the delivery of EVs are summarized in Table 3.
Peptide-guided delivery of cells
Enhancing the tumor homing of cytotoxic T lymphocytes (CTLs) in adoptive cell therapy is of high demand. Thus, chimeric antigen receptor (CAR)-T cells have been used to address this limitation. CAR-T cells are genetically engineered to express a chimeric receptor composed of an antibody against a tumor antigen (such as CD19), a cytoplasmic domain of the zeta chain of the T-cell receptor, and a costimulator domain42,43. In contrast, the nongenetic modification of the cell surface can reduce unexpected risks caused by genetic engineering of cells. CTLs labeled with the IL4R-binding IL4RPep-1 peptide (CRKRLDRNC) using a phospholipid-based membrane anchor showed enhanced tumor homing and antitumor growth activity in mice bearing B16F10 melanoma44. Apart from CTLs, mesenchymal stem cells (MSCs) conjugated with an E-selectin-targeting peptide (CGSDITWDQLWDLMK) on the cell surface showed controlled adhesion and rolling through an interaction between the peptide on the stem cells and E-selectin on the endothelial cells45. In addition, the nongenetic surface modification of MSCs with sialyl LewisX carbohydrate using a polyacrylamide linker and biotin/streptavidin interaction showed robust rolling on the endothelium and homed inflamed tissues in vivo more efficiently than unlabeled MSCs46.
Peptide-targeted cytotoxic peptides
Cationic amphipathic peptides with inherent cytotoxicity exhibit advantages: they can attenuate multidrug resistance in tumor cells and present broad-spectrum antitumor activities47,48. In contrast, they have drawbacks, including poor membrane permeability, suboptimal therapeutic activity, and structural instability48. A typical example is the KLAKLAKKLAKLAK or (KLAKLAK)2 proapoptotic peptide, which was originally developed as an antimicrobial peptide. In mammalian cells, it triggers mitochondrial membrane disruption and cytochrome C release, subsequently inducing cell apoptosis49,50. The (KLAKLAK)2 peptide encapsulated into mesoporous nanoparticles induced mitochondrial swelling and apoptosis51. Combining the (KLAKLAK)2 peptide with the CNGRC peptide, an aminopeptidase N-targeting peptide, efficiently inhibited tumor growth by targeting the enzyme present in angiogenic tumor endothelial cells52. The (KLAKLAK)2 peptide fused with the IL4R-binding IL4RPep-1 peptide (CRKRLDRNC) exhibited selective cytotoxicity toward IL4R-expressing tumor cells and enhanced the sensitivity of cells to chemotherapy53. The IL4R-targeted (KLAKLAK)2 peptide acted on IL-4R-high and M2-polarized TAMs as well as tumor cells and reduced the proportion of M2-type TAMs in the tumor microenvironment54. Moreover, the (KLAKLAK)2 peptide guided by CD44v6-binding (CNLNTIDTC and CNEWQLKSC), Her-2-binding (YCDGFYACYMDV), prostate tumor-targeting (SMSIARL), and bladder tumor-targeting (CSNRDARRC) peptides efficiently inhibited tumor growth with minimal effects on normal tissues55,56,57,58,59.
In addition, other cytotoxic or lytic peptides, such as defensin 1 (ACYCRIPACIAGERRYGTCIYQGRLWAFCC), cecropin B (KWKVFKKIEKMGRNIRNGIVKAGPAIAVLGEAKAL), magainins (GIGKFLHSAKKFGKAFVGEIMSNS), and dermaseptin (ALWKEVLKNAGKAALNEINNLVG), can increase membrane permeability and promote cell death60,61. The lactoferrin 5 derivative (PAWRKAFRWAWRMLKKAA) also showed selective cytotoxicity to tumor cells62. The eMTD peptide (KLNFRQKLLNLISKLFCSGT), consisting of the BH3 domain and mitochondrial targeting domain of the Noxa protein, causes cell membrane damage and necrotic cell death by interacting with voltage-dependent anion channel 263. Moreover, a peptide consisting of a prostate-specific membrane antigen (PSMA) substrate linked to a membrane-disrupting amoebapore H3 peptide (GFIATLCTKVLDFGIDKLQLIEDK) was highly active against PSMA-expressing LNCaP prostate cancer cells but not against PSMA-negative PC3 prostate cancer cells64. The cytotoxic peptides described in this section are summarized in Table 4.
Peptide-targeted radionuclides: Peptide receptor radionuclide therapy
Peptide receptor radionuclide therapy (PRRT) involves the combination of a tumor-homing peptide with a radionuclide or radioactive isotope as the therapeutic substance. The advantages of PRRT include its selectiveness in delivering radionuclides, which reduces systemic side effects, and its effective control of advanced, inoperable or metastatic tumors; however, radiation-induced toxicity to healthy organs, especially the bone marrow, remains a major limitation65. Octreotide (Sandostatin®, FCFWKTCT), an 8-mer peptide of somatostatin analog, plays a vital role in treating patients with neuroendocrine tumors66. PRRT with octreotide aims to selectively irradiate somatostatin receptor 2 (SSTR2)-expressing neuroendocrine tumor cells and the surrounding blood vessels to inhibit the angiogenetic response during treatment67. 111In is linked to octreotide using diethylenetriamine pentaacetic acid, while 90Y and 177Lu (Lutathera®) are linked using tetraazacyclododecane tetraacetic acid as a chelator68. In addition to SSTR2, PRRT has been extended to other receptors, such as the gastrin-releasing peptide (GRP) and cholecystokinin-2 (CCK-2) receptors. 99mTc-conjugated RP527 peptide (VPLPAGGGTVLTKMYPRGNHWAVGHLM), a GRP analog, has been exploited for treating human malignancies, including colon and prostate carcinomas69. 111In-labeled minigastrin (LEEEEEAYGWMDF), a CCK-2 receptor-selective peptide, has been employed to treat human colorectal and pancreatic tumors70.
Peptide-targeted small-molecule drugs: peptide–drug conjugates
Peptide–drug conjugates (PDCs) comprise three elements: a tumor-homing peptide, linker, and cytotoxic agent (Fig. 2). Small molecule-based cytotoxic agents have advantages of high oral availability, metabolic stability, and high membrane permeability, while having disadvantages of high toxicity, poor solubility, and lower selectivity than alternatives71. The delivery of PDCs into tumor cells via tumor-homing peptides can exert a tumoricidal effect in the intracellular compartments of tumor cells where tumor-specific pH or enzymes can break the linkers, releasing the drugs. Considering that PDCs increased the local concentration of cytotoxic agents in tumor tissues, they can reduce cytotoxic effects to normal tissues and increase therapeutic efficacy. For antibody–drug conjugates (ADCs), the market size in terms of revenue is predicted to exceed 16 billion dollars by 202672. Compared with ADCs, PDCs exhibit better tumor penetration because of their small molecular weight, lower systemic exposure (owing to rapid clearance from the body), lower risk of immunogenicity and liver damage, and easier and cheaper production methods.

PDCs comprise a tumor-homing peptide, linker, and cytotoxic agent. The linkers used for PDCs are cleaved by intracellular enzymes or the acidic pH environment inside tumor cells, whereas some linkers are noncleavable. Small molecules are commonly used as cytotoxic agents for PDCs, and in certain cases, bacterial toxins are used.
Diverse linkers have been designed to conjugate drugs or cytotoxic agents with tumor-homing peptides73,74. Selecting an appropriate linker is crucial for designing PDCs. Furthermore, the microenvironment where PDCs function should be considered because linkers impact drug efficacy or binding affinity depending on structural differences of the linkers. For example, certain types of peptide linkers are designed to be cleaved by enzymes abundant in tumor cells to selectively release drugs to these cells. These linkers include the GFLG peptide which is cleaved by cathepsin B75, the PLGLAG peptide which is cleaved by matrix metalloprotease (MMP)-2/976, and the oxime-hydrazone bond which is hydrolyzed in an acidic pH77.
The SSTR2-binding octreotide was conjugated to doxorubicin via a cleavable disulfide bond and used for the treatment of pituitary, pancreatic, and breast tumors78. The disulfide bonds can be cleaved by reduced glutathione (GSH) in cells. The αvβ3 integrin-binding RGD4C peptide was conjugated to PD0325901, an MEK1/2 inhibitor, via a GGGGG peptide linker, which enhanced the antitumor activity of the drug against glioblastoma79. The RGDfK peptide-camptothecin conjugate linked by a Lys splitter enhanced cytotoxicity to melanoma and non-small cell lung cancer cells80. EGFR-binding GE11 peptide was linked to doxorubicin via the disulfide bond and used for hepatocellular tumors81. The angiopep-2 peptide that binds to low-density lipoprotein receptor-related protein-1 (LRP-1) was conjugated to paclitaxel via a succinyl group (named ANG1005) and applied to the treatment of glioma and metastatic breast cancer82. The tumor-homing peptides and linkers involved in the generation of PDCs are summarized in Table 5. Several PDCs are being considered for approval by the Food and Drug Administration (FDA) for commercial use. For example, BT8009, comprising a bicyclic peptide (CP(1Nal)dCM(hArg)DWSTP(HyP)WC) as a targeting moiety and monomethyl auristatin (MMAE) as a cargo, targets Nectin-4 on tumor cells. This PDC is in phase I/II clinical trials for the treatment of patients with metastatic non-small cell lung cancer. The PDCs currently under clinical/preclinical trials are summarized in Table 6.
Peptide inhibitors or antagonists of cell surface proteins
Immune checkpoint inhibitors
The advent of immune checkpoint inhibitors (ICIs) has revolutionized the field of tumor therapy and promoted the development of more immune checkpoint blockades83. ICIs work by blocking the interactions between immune checkpoints such as CTL-associated protein 4 (CTLA-4), programmed cell death-1 (PD-1), and programmed cell death ligand-1 (PD-L1) and their ligands, which releases the inhibitory brakes of T cells and results in the robust activation of immune responses (Fig. 3). For example, CTLA-4, an inhibitory receptor expressed primarily by T cells, dampens T-cell activity and is upregulated upon T-cell activation84,85. At present, ICIs are used as first-line therapies for various solid tumors. Over the past decades, antibodies have been widely used ICIs. Ipilimumab was the first CTLA-4-blocking antibody approved by the US FDA for the treatment of human cancers. Anti-PD-1 antibodies such as pembrolizumab and nivolumab were included the second set of antibodies to be approved for the treatment of human malignancies, followed by anti-PD-L1 antibodies such as atezolizumab, durvalumab, and avelumab86.

Interactions between immune checkpoints, such as CTLA-4 and PD-1 on T cells as well as CD80/CD86 and PD-L1 on antigen presenting and tumor cells, respectively, suppress T-cell activity. In addition, TIGIT, TIM3, and LAG3 on T cells play roles as immune checkpoints by interacting with their partners, such as CD155, galectin 9, and major histocompatibility complex, respectively. Blue arrows represent stimulatory signals, while red lines represent inhibitory signals.
PD-L1 is frequently upregulated in the tumor cell microenvironment as well as in DCs, macrophages, myeloid-derived suppressor cells (MDSCs), and regulatory T cells87,88,89,90. PD-L1 interacts with its ligand PD-1. Although T cells recognize tumor cells in the human body and kill them, the interaction between PD-1 on T cells and PD-L1 on tumor cells leads to T cell exhaustion91,92,93. Peptides that can block the PD-1/PD-L1 interaction and restore T cell activity against tumor cells have been identified94,95,96,97,98,99,100,101; these include peptide-57 (F(NMeAla)NPHLSWSW(NMeNle)(NMeNle)RCG), CLP001 (HYPERPHANQAS)/CLP002 (WHRSYYTWNLNT), and PD-L1Pep-1 (CLQKTPKQC)/PD-L1Pep-2 (CVRARTR) peptides. In addition to inducing T-cell reinvigoration through their PD-L1-blocking activity, PD-L1-binding peptides enable the targeted delivery of chemotherapeutic drugs to PD-L1-high tumors using PD-L1 as a tumor target. For example, PD-L1Pep-2 peptide-labeled doxorubicin-loaded liposomes increased the CD8 + T-cell/regulatory T-cell ratio in mouse colon tumor tissues more efficiently than combined treatment with PD-L1Pep-2 peptide and untargeted doxorubicin-loaded liposomes101. A prodrug nanoparticle synthesized by conjugating PD-L1Pep-2 with doxorubicin via cathepsin B-cleavable peptide linker (FRRG) inhibited tumor progression in the 4T1 mouse breast tumor model by inducing doxorubicin-mediated immunogenic cell death and blocking PD-L1 through PD-L1Pep-2102. Moreover, labeling peptides in nanoparticles increases the binding affinity of the peptide. For example, ferritin nanocages with multivalent PD-L1Pep-1 peptide bound to PD-L1 with a higher affinity than free PD-L1Pep-1 (~30 nM vs. 300 nM)103.
Recently, peptides that target next-generation immune checkpoints, such as T-cell immunoglobulin-3 (TIM-3), lymphocyte activation gene 3 (LAG-3), and T-cell immunoreceptor with Ig and ITIM domains (TIGIT), have attracted increasing attention. A TIM-3-binding peptide (GLIPLTTMHIGK) interferes with the binding of TIM-3 to Gal-9, the main ligand of TIM-3, thereby enhancing T-cell activity. Combining this peptide with a PD-L1 inhibitor exerted a tumor-suppressive effect in a mouse model104. A disulfide-bound cyclic LAG-3-binding peptide (CVPMTYRAC) interfered with the binding of LAG-3 to HLA-DR, the main ligand of LAG-3, activating CD8 + T cells while reducing the proportion of regulatory T cells105. A D-form version of a TIGIT-binding peptide (GGYTFHWHRLNP) identified from mirror-image phage display exhibited proteolytic resistance and prolonged half-life; it blocked the binding area of TIGIT to the poliovirus receptor (or CD155), enhanced the function of CD8 + T cells, and inhibited tumor growth106. The peptides that block immune checkpoints are summarized in Table 7.
Peptide antagonists of receptor tyrosine kinases, kinase-associated receptors, and other surface proteins
Tumor cells express abundant cell surface receptors for growth factors. Thus, receptor blockers or antagonistic antibodies and peptides can be used as anticancer agents. c-Met is a receptor tyrosine kinase that is overexpressed in numerous tumors. It binds to hepatocyte growth factor (HGF) and plays an important role in tumorigenesis and metastasis107. Using computer simulation, novel sequences of peptides, including the CM7 peptide (DQIIANN), have been designed to bind to c-Met with high affinity. This novel peptide bound to c-Met-expressing cells, inhibiting c-Met-mediated cell migration and invasion and tumor progression in mice108. A disulfide-constrained HGF-binding peptide, namely, HB10 (VNWVCFRDVGCDWVL), inhibits HGF–c-Met binding109. Soluble heparin-binding epidermal growth factor (sHB-EGF) is another target in combating cancer tumorigenesis and metastasis. Two sHB-EGF-binding peptides, namely, DRWVARDPASIF and TVGLPMTYYMHT, have been identified using phage display. They suppressed the activity of sHB-EGF to promote ovarian tumor cell migration and invasion by inhibiting the EGFR signaling pathway110.
CD44 is a cell surface receptor involved in cell adhesion to the extracellular matrix111. Although CD44 is expressed in normal cells, its alternative splicing isoforms, including CD44 variant 6 (CD44v6), are upregulated in tumor cells, contributing to tumor cell migration and metastasis by interacting with c-Met111. Using structural analysis, v6pep (KEQWFGNRWHEGYR) was selected from the human CD44v6 domain that interacts with c-Met and inhibits tumor growth and metastasis in a pancreatic cancer model112,113. Presently, v6pep is undergoing clinical trials. By screening a phage-displayed random peptide library, the NLN (CNLNTIDTC) and NEW (CNEWQKLSC) peptides that bind to CD44v6-expressing cells were selected; these peptides hindered HGF-mediated c-Met activation, thereby inhibiting CD44v6-high tumor cell migration and invasion55.
Certain tumor-derived exosomes contain heat shock protein 72 (Hsp72) in their membrane and interact with Toll-like receptor 2 (TLR2) on MDSCs, thereby activating cells114. The A8 peptide (SPWPRPTY) blocked the interaction between Hsp72 and TLR2 and the subsequent activation of MDSCs, thereby inhibiting tumor progression and potentiating the antitumor effect of chemotherapeutic agents, such as cisplatin115. Thus, peptides that act as cell surface protein antagonists are potential tools for inhibiting tumor progression and metastasis and can be administered alone or in combination with chemotherapy. The peptides that block cell surface receptors described here are summarized in Table 8.
Peptide antagonists of hormone receptors
Some cancers depend on hormones to grow; thus, blocking the action of hormones can slow or control cancer growth. This kind of therapy is known as hormone therapy or endocrine therapy. At present, hormone therapy is applied to certain kinds of cancers, such as breast and prostate cancers. Hormone therapy, when used before surgery or radiation therapy as an adjuvant therapy, can decrease tumor size and lower the risk for tumor recurrence.
Gonadotrophin-releasing hormone (GnRH), also known as luteinizing hormone-releasing hormone, is released from the hypothalamus. It binds to a GnRH receptor in the pituitary to increase the production of follicle-stimulating and luteinizing hormones, thereby stimulating the release of estrogen by the ovaries116. When a GnRH analog is first administered, it produces a surge in ovarian hormones that can also cause several adverse effects, such as hot flashes. However, the long-term administration of the GnRH analog reduces ovarian hormone production and secretion, which downregulates and desensitizes the GnRH receptor in pituitary gonadotropic cells116. The GnRH receptor is also found in certain cancers, and the reduction in circulating estrogen slows the growth of hormone receptor-positive tumors such as ovarian cancer117, prostate cancer118, and breast cancer119,120,121. The use of GnRH analogs in clinical settings has been complicated because of their short half-life. However, with some modifications in its amino acids, long-lasting analogs have been successfully developed and used in the treatment of breast and prostate cancers. GnRH analogs that are currently used in clinics include goserelin (Zoladex®), (pGlu)HWSY(D-Ser(But)LRP), leuprorelin or leuprolide (Lupron®, (pGlu)HWSY(D-Leu)LRP), and triptorelin (Decapeptyl®, (pGlu)HWSY(D-Trp)LRPG).
Somatostatin (AGCKNFFWKTFTSC) is a peptide produced by paracrine cells located throughout the gastrointestinal tract and binds to somatostatin receptors (SSTRs). Octreotide (FCFWKTCT) is a somatostatin analog that binds to SSTR2 and SSTR5 and serves as a growth hormone, insulin, and glucagon inhibitor122. Octreotide is used to treat severe diarrhea caused by certain intestinal tumors, such as vasoactive intestinal peptide-secreting tumors or metastatic carcinoid tumors.
Peptide inhibitors of intracellular protein–protein interactions
Intracellular protein–protein interactions (PPIs) play a critical role in cells; for example, they facilitate the formation of protein complexes for signal transduction and facilitate the binding of transcription factors to promoters and enhancers. Thus, pharmacological approaches have been exploited to inhibit intracellular PPIs; related compounds include small molecules based on chemicals with a molecular weight <500 Da and biologicals based on proteins with a molecular weight >5000 Da. Small molecules efficiently cross the cell membrane, and they regulate the action of intracellular proteins123. However, these drugs cannot recognize a single mutation at the target site, and tumor cells easily acquire resistance against these drugs. In addition, the large surface of proteins involved in the interaction among proteins is not covered by small molecules because their sizes are too small124. In contrast, biologicals can bind to larger interfaces of proteins with high selectivity. However, they have poor cell permeability125. In addressing the limitations of small molecules and biologicals, peptides that interfere with PPIs with a molecular weight ranging between 500 and 5000 Da have been developed126. Peptides have the benefits of small molecules and biologicals, including the cell permeability of small molecules and the high selectivity of biologicals covering a large surface of proteins127. Considering that the sequence of peptide inhibitors frequently originates from endogenous proteins involved in the interaction, most of them serve as competitors of native protein interactions128.
c-Myc is a transcription factor involved in diverse human malignant tumors129,130. It usually forms heterodimeric complexes with its partner transcription factors to bind to DNA and regulate gene expression131. A peptide comprising 14 amino acids (RQIKIWFQNRRMKWKK) that originated from the helix 1 C-terminal region of Myc blocks the interaction between c-Myc and its partner132,133. Another example is OmoMyc, which comprises 92 amino acids and originates from the bHLHZip region of Myc but differs from Myc in four amino acid residues134,135,136.
Homeobox (HOX) is an important transcription factor for body segmentation and patterning during vertebrate development137. HOX gene expression is generally enhanced in tumors and is associated with angiogenesis, metastasis, and proliferation of tumor cells138,139. A common cofactor of HOX is preB-cell leukemia homeobox (PBX)140,141. The HXR9 peptide (WYPWMKKHHRRRRRRRRR) interferes with the interactions between HOX and PBX in several mouse tumor models139,142.
KRAS is an oncogenic protein that is commonly activated in many tumors, including lung cancer and pancreatic cancer, and it has been considered an undruggable target because it lacks a classical drug binding site143,144. The KRpep-2d peptide (Ac-RRRR-cyclo(CPLYISYDPVC)-NH2), a macrocyclic peptide that is a cyclic peptide containing >12 amino acids, and its derivatives bind to KRAS and inhibit KRAS-downstream signaling and cell proliferation145,146.
Perspectives: improvement of the pharmacokinetic properties and biological activity of peptides
Several approaches have been exploited to address or reduce the drawbacks of peptides as therapeutics (Fig. 4). First, to increase resistance to degradation, peptides are chemically modified through cyclization, which involves formation of disulfide bonds or formation of a stapled peptide; through N-term acetylation or C-term amidation; through modification to D-form amino acids; and through replacement of amino acids with unnatural amino acids or peptoids. Second, to slow down the excretion out of the body and increase half-life in the blood, peptides can be fused with the Fc fragment of an antibody and protein scaffolds such as Staphylococcus A antigen (Affibody) or conjugated with polyethylene glycol and fatty acids to enables the peptides to bind to albumin. Third, multivalent labeling of tumor-homing peptides on drug-loaded nanoparticles, EVs, and cells can enhance the binding activity and stability of peptides. Fourth, a peptide that binds to an intracellular target protein can be combined with a peptide that binds to an E3 ligase to degrade the target protein via proteolysis targeting chimera (ProTac) technology. Such peptide-based ProTacs have already been reported147,148. Fifth, tumor-homing peptides are linked with chemotherapeutic drugs to increase the antitumor activity of peptides. Finally, peptides are loaded into long-acting release microspheres or depots and injected into tissues to slowly release peptides for a longer time.

Protease-mediated degradation and renal clearance of peptides can be reduced via chemical modifications. The long-acting release of peptides can be obtained by certain formulations, such as “depots”. Peptide-based ProTac, peptide–drug conjugates, and multivalent labeling on nanoparticles can improve the pharmacokinetic properties and biological activity of peptides.
At present, a major portion of peptide therapeutics in the clinic are diabetes drugs such as liraglutide and dulaglutide. In the current market, peptide-based anticancer therapeutics include hormone analogs such as octreotide, leuprolide, and goserelin. In the future, an increasing number of peptide therapeutics will be developed in the field of cancer therapy; these could include tumor-homing peptides for targeted delivery of nanoparticles or EVs, peptide antagonists against cell surface proteins, and interference peptides against PPIs. In addition, PDCs could be used as an alternative to ADCs for certain cancers. Moreover, peptide-based ProTac technology will address the resistance of tumor cells to chemotherapy and will be a potential tool for cancer therapy.
References
Ladner, R. C., Sato, A. K., Gorzelany, J. & de Souza, M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Disco. 9, 525–529 (2004).
Fosgerau, K. & Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Disco. 20, 122–128 (2015).
Blanco‐Míguez, A. et al. From amino acid sequence to bioactivity: the biomedical potential of antitumor peptides. Protein Sci. 25, 1084–1095 (2016).
Wang, L. et al. Therapeutic peptides: current applications and future directions. Signal Transduct. Target Ther. 7, 48 (2022).
Kessel, C. et al. Multimerization of peptide mimotopes for blocking of factor VIII neutralizing antibodies. Chem. Med. Chem. 4, 1364–1370 (2009).
Knittelfelder, R., Riemer, A. B. & Jensen-Jarolim, E. Mimotope vaccination–from allergy to cancer. Expert Opin. Biol. Ther. 9, 493–506 (2009).
Tong, A. H. Y. et al. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295, 321–324 (2002).
Liu, M., Fang, X., Yang, Y. & Wang, C. Peptide-enabled targeted delivery systems for therapeutic applications. Front Bioeng. Biotechnol. 9, 701504 (2021).
Chen, Y. et al. Sortase A-mediated cyclization of novel polycyclic RGD peptides for ανβ3 integrin targeting. Bioorg. Med Chem. Lett. 73, 128888 (2022).
Echigo, H. et al. Development and evaluation of a theranostic probe with RGD peptide introduced platinum complex to enable tumor-specific accumulation. Bioorg. Med. Chem. 70, 116919 (2022).
Kato, N. et al. Synthesis and evaluation of a novel adapter lipid derivative for preparation of cyclic peptide-modified PEGylated liposomes: Application of cyclic RGD peptide. Eur. J. Pharm. Sci. 176, 106239 (2022).
Chen, K. & Chen, X. Integrin targeted delivery of chemotherapeutics. Theranostics 1, 189 (2011).
Sugahara, K. N. et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer cell 16, 510–520 (2009).
Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 110, 3–12 (2017).
Teesalu, T., Sugahara, K. N., Kotamraju, V. R. & Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl Acad. Sci. USA 106, 16157–16162 (2009).
Chen, T. et al. nRGD modified lycobetaine and octreotide combination delivery system to overcome multiple barriers and enhance anti-glioma efficacy. Colloids Surf. B 156, 330–339 (2017).
Su, S., Wang, H., Liu, X., Wu, Y. & Nie, G. iRGD-coupled responsive fluorescent nanogel for targeted drug delivery. Biomaterials 34, 3523–3533 (2013).
Yu, K.-F. et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J. Nanomed. 8, 2473 (2013).
Fogal, V., Zhang, L., Krajewski, S. & Ruoslahti, E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 68, 7210–7218 (2008).
Karmali, P. P. et al. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine 5, 73–82 (2009).
Hardwick, J. S. et al. Identification of biomarkers for tumor endothelial cell proliferation through gene expression profiling. Mol. Cancer Ther. 4, 413–425 (2005).
Yu, D.-H., Lu, Q., Xie, J., Fang, C. & Chen, H.-Z. Peptide-conjugated biodegradable nanoparticles as a carrier to target paclitaxel to tumor neovasculature. Biomaterials 31, 2278–2292 (2010).
Obiri, N., Siegel, J., Varricchio, F. & Puri, R. Expression of high-affinity IL-4 receptors on human melanoma, ovarian and breast carcinoma cells. Clin. Exp. Immunol. 95, 148–155 (1994).
Kawakami, K., Leland, P. & Puri, R. K. Structure, function, and targeting of interleukin 4 receptors on human head and neck cancer cells. Cancer Res. 60, 2981–2987 (2000).
Kawakami, M. et al. Interleukin 4 receptor on human lung cancer: a molecular target for cytotoxin therapy. Clin. Cancer Res. 8, 3503–3511 (2002).
Kim, J.-H. et al. Facilitated intracellular delivery of peptide-guided nanoparticles in tumor tissues. J. Control Release 157, 493–499 (2012).
Chi, L. et al. Enhanced delivery of liposomes to lung tumor through targeting interleukin-4 receptor on both tumor cells and tumor endothelial cells. J. Control Release 209, 327–336 (2015).
Jeon, J. O. et al. Designed nanocage displaying ligand-specific peptide bunches for high affinity and biological activity. ACS Nano 7, 7462–7471 (2013).
Guruprasath, P. et al. Interleukin-4 receptor-targeted delivery of Bcl-xL siRNA sensitizes tumors to chemotherapy and inhibits tumor growth. Biomaterials 142, 101–111 (2017).
Namgung, R. et al. Poly-cyclodextrin and poly-paclitaxel nano-assembly for anticancer therapy. Nat. Commun. 5, 1–12 (2014).
de Groot, A. E. et al. Targeting interleukin 4 receptor alpha on tumor-associated macrophages reduces the pro-tumor macrophage phenotype. Neoplasia 32, 100830 (2022).
Bankaitis, K. V. & Fingleton, B. Targeting IL4/IL4R for the treatment of epithelial cancer metastasis. Clin. Exp. Metastasis 32, 847–856 (2015).
Figueiredo, P. et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomater. 133, 231–243 (2021).
Johnsen, K. B. et al. A comprehensive overview of exosomes as drug delivery vehicles—endogenous nanocarriers for targeted cancer therapy. Biochim Biophys. Acta Rev. Cancer 1846, 75–87 (2014).
Alvarez-Erviti, L. et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345 (2011).
Tian, Y. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 35, 2383–2390 (2014).
Ohno, S.-i et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 21, 185–191 (2013).
Morishita, M., Takahashi, Y., Nishikawa, M., Ariizumi, R. & Takakura, Y. Enhanced class I tumor antigen presentation via cytosolic delivery of exosomal cargos by tumor-cell-derived exosomes displaying a pH-sensitive fusogenic peptide. Mol. Pharm. 14, 4079–4086 (2017).
Gunassekaran, G. R., Vadevoo, S. M. P., Baek, M.-C. & Lee, B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials 278, 121137 (2021).
Zhan, Q. et al. Engineering blood exosomes for tumor-targeting efficient gene/chemo combination therapy. Theranostics 10, 7889 (2020).
Cheng, H. et al. Chimeric peptide engineered exosomes for dual-stage light guided plasma membrane and nucleus targeted photodynamic therapy. Biomaterials 211, 14–24 (2019).
Lim, W. A. & June, C. H. The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017).
Maus, M. V. & June, C. H. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin. Cancer Res. 22, 1875–1884 (2016).
Gunassekaran, G. R. et al. Non-genetic engineering of cytotoxic T cells to target IL-4 receptor enhances tumor homing and therapeutic efficacy against melanoma. Biomaterials 159, 161–173 (2018).
Cheng, H. et al. Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell–selectin interaction kinetics. Biomaterials 33, 5004–5012 (2012).
Sarkar, D. et al. Engineered cell homing. Blood 118, e184–e191 (2011).
Johnstone, S. A., Gelmon, K., Mayer, L. D., Hancock, R. E. & Bally, M. B. In vitro characterization of the anticancer activity of membrane-active cationic peptides. I. Peptide-mediated cytotoxicity and peptide-enhanced cytotoxic activity of doxorubicin against wild-type and p-glycoprotein over-expressing tumor cell lines. Anticancer Drug Des. 15, 151–160 (2000).
Luan, X. et al. Cytotoxic and antitumor peptides as novel chemotherapeutics. Nat. Prod. Rep. 38, 7–17 (2021).
Mai, J. C., Mi, Z., Kim, S.-H., Ng, B. & Robbins, P. D. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 61, 7709–7712 (2001).
Dufort, S. et al. Targeted delivery of a proapoptotic peptide to tumors in vivo. J. Drug Target 19, 582–588 (2011).
Jin, Y. et al. Nanosystem composed with MSNs, gadolinium, liposome and cytotoxic peptides for tumor theranostics. Colloids Surf. B 151, 240–248 (2017).
Ellerby, H. M. et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med 5, 1032–1038 (1999).
Permpoon, U. et al. Inhibition of tumor growth against chemoresistant cholangiocarcinoma by a proapoptotic peptide targeting interleukin-4 receptor. Mol. Pharm. 17, 4077–4088 (2020).
Vadevoo, S. M. P. et al. IL4 receptor–targeted proapoptotic peptide blocks tumor growth and metastasis by enhancing antitumor immunity anticancer activity of IL4R-targeted proapoptotic peptide. Mol. Cancer Ther. 16, 2803–2816 (2017).
Khan, F. et al. Identification of novel CD44v6-binding peptides that block CD44v6 and deliver a pro-apoptotic peptide to tumors to inhibit tumor growth and metastasis in mice. Theranostics 11, 1326 (2021).
Fantin, V. R. et al. A bifunctional targeted peptide that blocks HER-2 tyrosine kinase and disables mitochondrial function in HER-2-positive carcinoma cells. Cancer Res. 65, 6891–6900 (2005).
Arap, W. et al. Targeting the prostate for destruction through a vascular address. Proc. Natl Acad. Sci. USA 99, 1527–1531 (2002).
Jung, H.-K. et al. Bladder tumor-targeted delivery of pro-apoptotic peptide for cancer therapy. J. Control Release 235, 259–267 (2016).
Vadevoo, S. M. P. et al. Peptide-based targeted therapeutics and apoptosis imaging probes for cancer therapy. Arch. Pharm. Res. 42, 150–158 (2019).
Yamasaki, K. & Gallo, R. L. Antimicrobial peptides in human skin disease. Eur. J. Dermatol 18, 11–21 (2008).
Leuschner, C. & Hansel, W. Membrane disrupting lytic peptides for cancer treatments. Curr. Pharm. Des. 10, 2299–2310 (2004).
Yang, N., Lejon, T. & Rekdal, Ø. Antitumour activity and specificity as a function of substitutions in the lipophilic sector of helical lactoferrin‐derived peptide. J. Pept. Sci. 9, 300–311 (2003).
Han, J.-H. et al. Noxa mitochondrial targeting domain induces necrosis via VDAC2 and mitochondrial catastrophe. Cell Death Dis. 10, 1–13 (2019).
Warren, P. et al. In vitro targeted killing of prostate tumor cells by a synthetic amoebapore helix 3 peptide modified with two γ-linked glutamate residues at the COOH terminus. Cancer Res. 61, 6783–6787 (2001).
Ersahin, D., Doddamane, I. & Cheng, D. Targeted radionuclide therapy. Cancers 3, 3838–3855 (2011).
Costa, F. & Gumz, B. Octreotide–a review of its use in treating neuroendocrine tumours. Eur. Endocrinol. 10, 70 (2014).
Bodei, L., Pepe, G. & Paganelli, G. Peptide receptor radionuclide therapy (PRRT) of neuroendocrine tumors with somatostatin analogues. Eur. Rev. Med Pharm. Sci. 14, 347–351 (2010).
Esser, J.-P. et al. Comparison of [177Lu-DOTA0, Tyr3] octreotate and [177Lu-DOTA0, Tyr3] octreotide: which peptide is preferable for PRRT? Eur. J. Nucl. Med. Mol. Imaging 33, 1346–1351 (2006).
Van de Wiele, C. et al. Biodistribution and dosimetry of 99mTc-RP527, a gastrin-releasing peptide (GRP) agonist for the visualization of GRP receptor-expressing malignancies. J. Nucl. Med. 42, 1722–1727 (2001).
Laverman, P. et al. Targeting of a CCK2 receptor splice variant with 111In-labelled cholecystokinin-8 (CCK8) and 111In-labelled minigastrin. Eur. J. Nucl. Med. Mol. Imaging 35, 386–392 (2008).
La Manna, S., Di Natale, C., Florio, D. & Marasco, D. Peptides as therapeutic agents for inflammatory-related diseases. Int J. Mol. Sci. 19, 2714 (2018).
do Pazo, C., Nawaz, K. & Webster, R. M. The oncology market for antibody-drug conjugates. Nat. Rev. Drug Discov. 20, 583–584 (2021).
Hoppenz, P., Els-Heindl, S. & Beck-Sickinger, A. G. Peptide-drug conjugates and their targets in advanced cancer therapies. Front Chem. 8, 571 (2020).
Chavda, V. P., Solanki, H. K., Davidson, M., Apostolopoulos, V. & Bojarska, J. Peptide-drug conjugates: a new hope for cancer management. Molecules 27, 7232 (2022).
Raza Naqvi, S. A. et al. Insertion of a lysosomal enzyme cleavage site into the sequence of a radiolabeled neuropeptide influences cell trafficking in vitro and in vivo. Cancer Biother Radiopharm. 25, 89–95 (2010).
van Duijnhoven, S. M., Robillard, M. S., Nicolay, K. & Grüll, H. Tumor targeting of MMP-2/9 activatable cell-penetrating imaging probes is caused by tumor-independent activation. J. Nucl. Med. 52, 279–286 (2011).
Szabó, I. et al. Development of an oxime bond containing daunorubicin-gonadotropin-releasing hormone-III conjugate as a potential anticancer drug. Bioconjug Chem. 20, 656–665 (2009).
Lelle, M. et al. Octreotide-mediated tumor-targeted drug delivery via a cleavable doxorubicin–peptide conjugate. Mol. Pharm. 12, 4290–4300 (2015).
Hou, J. et al. RGD peptide conjugation results in enhanced antitumor activity of PD0325901 against glioblastoma by both tumor-targeting delivery and combination therapy. Int J. Pharm. 505, 329–340 (2016).
Gilad, Y. et al. Dual‐drug RGD conjugates provide enhanced cytotoxicity to melanoma and non‐small lung cancer cells. Pept. Sci. 106, 160–171 (2016).
Fan, M. et al. Design and biological activity of epidermal growth factor receptor-targeted peptide doxorubicin conjugate. Biomed. Pharmacother. 70, 268–273 (2015).
Regina, A. et al. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep‐2. Br. J. Pharm. 155, 185–197 (2008).
Topalian, S. L., Taube, J. M. & Pardoll, D. M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 367, eaax0182 (2020).
Valk, E., Rudd, C. E. & Schneider, H. CTLA-4 trafficking and surface expression. Trends Immunol. 29, 272–279 (2008).
Krummel, M. F. & Allison, J. P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182, 459–465 (1995).
Bagchi, S., Yuan, R. & Engleman, E. G. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev. Pathol. 16, 223–249 (2021).
Zou, W., Wolchok, J. D. & Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med 8, 328rv324–328rv324 (2016).
Yi, M., Niu, M., Xu, L., Luo, S. & Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 14, 1–13 (2021).
Dong, P., Xiong, Y., Yue, J., Hanley, S. J. & Watari, H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front Oncol. 8, 386 (2018).
Nguyen, L. T. & Ohashi, P. S. Clinical blockade of PD1 and LAG3—potential mechanisms of action. Nat. Rev. Immunol. 15, 45–56 (2015).
Topalian, S. L., Taube, J. M., Anders, R. A. & Pardoll, D. M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287 (2016).
Tremblay-LeMay, R., Rastgoo, N. & Chang, H. Modulating PD-L1 expression in multiple myeloma: an alternative strategy to target the PD-1/PD-L1 pathway. J. Hematol. Oncol. 11, 1–16 (2018).
Li, X.-S., Li, J.-W., Li, H. & Jiang, T. Prognostic value of programmed cell death ligand 1 (PD-L1) for hepatocellular carcinoma: a meta-analysis. Biosci. Rep. 40, BSR20200459 (2020).
Surmiak, E. et al. PD-L1 inhibitors: different classes, activities, and mechanisms of action. Int J. Mol. Sci. 22, 11797 (2021).
Li, C. et al. Peptide blocking of PD-1/PD-L1 interaction for cancer immunotherapy effect of PD-L1–targeting peptide on cancer immunotherapy. Cancer Immunol. Res. 6, 178–188 (2018).
Magiera‐Mularz, K. et al. Bioactive macrocyclic inhibitors of the PD‐1/PD‐L1 immune checkpoint. Angew. Chem. Int Ed. Engl. 56, 13732–13735 (2017).
Caldwell, C. et al. Identification and validation of a PD-L1 binding peptide for determination of PDL1 expression in tumors. Sci. Rep. 7, 1–11 (2017).
Liu, H. et al. Discovery of low-molecular weight anti-PD-L1 peptides for cancer immunotherapy. J. Immunother. Cancer 7, 1–14 (2019).
Chen, Y. et al. Engineering a high-affinity PD-1 peptide for optimized Immune cell–mediated tumor therapy. Cancer Res. Treat. 54, 362 (2022).
Abbas, A. B. et al. Design and synthesis of A PD-1 binding peptide and evaluation of its anti-tumor activity. Int J. Mol. Sci. 20, 572 (2019).
Gurung, S. et al. Phage display-identified PD-L1-binding peptides reinvigorate T-cell activity and inhibit tumor progression. Biomaterials 247, 119984 (2020).
Moon, Y. et al. Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death. Theranostics 12, 1999 (2022).
Jeon, I. S. et al. Anticancer nanocage platforms for combined immunotherapy designed to harness immune checkpoints and deliver anticancer drugs. Biomaterials 270, 120685 (2021).
Zhong, T. et al. The biologically functional identification of a novel TIM3-binding peptide P26 in vitro and in vivo. Cancer Chemother. Pharm. 86, 783–792 (2020).
Zhai, W. et al. A novel cyclic peptide targeting LAG-3 for cancer immunotherapy by activating antigen-specific CD8+ T cell responses. Acta Pharm. Sin. B 10, 1047–1060 (2020).
Zhou, X. et al. A novel d‐peptide identified by mirror‐image phage display blocks TIGIT/PVR for cancer immunotherapy. Angew. Chem. Int Ed999 59, 15114–15118 (2020).
Ma, P. C., Maulik, G., Christensen, J. & Salgia, R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 22, 309–325 (2003).
Xia, C. et al. Novel Peptide CM 7 Targeted c-met with antitumor activity. Molecules 25, 451 (2020).
Tam, E. M. et al. Noncompetitive inhibition of hepatocyte growth factor-dependent Met signaling by a phage-derived peptide. J. Mol. Biol. 385, 79–90 (2009).
Shen, Y. et al. Discovery of HB-EGF binding peptides and their functional characterization in ovarian cancer cell lines. Cell Death Disco. 5, 1–11 (2019).
Yan, Y., Zuo, X. & Wei, D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl. Med 4, 1033–1043 (2015).
Matzke, A., Herrlich, P., Ponta, H. & Orian-Rousseau, V. A five-amino-acid peptide blocks Met-and Ron-dependent cell migration. Cancer Res. 65, 6105–6110 (2005).
Matzke-Ogi, A. et al. Inhibition of tumor growth and metastasis in pancreatic cancer models by interference with CD44v6 signaling. Gastroenterol 150, 513–525. e510 (2016).
Chalmin, F. et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest 120, 457–471 (2010).
Gobbo, J. et al. Restoring anticancer immune response by targeting tumor-derived exosomes with a HSP70 peptide aptamer. J. Natl Cancer Inst. 108, djv330 (2016).
Stamatiades, G. A. & Kaiser, U. B. Gonadotropin regulation by pulsatile GnRH: signaling and gene expression. Mol. Cell Endocrinol. 463, 131–141 (2018).
Srkalovic, G., Wittliff, J. L. & Schally, A. V. Detection and partial characterization of receptors for [D-Trp6]-luteinizing hormone-releasing hormone and epidermal growth factor in human endometrial carcinoma. Cancer Res. 50, 1841–1846 (1990).
Tieva, Å., Stattin, P., Wikström, P., Bergh, A. & Damber, J. E. Gonadotropin‐releasing hormone receptor expression in the human prostate. Prostate 47, 276–284 (2001).
Huerta-Reyes, M. et al. Treatment of breast cancer with gonadotropin-releasing hormone analogs. Front Oncol. 9, 943 (2019).
Lu, Y.-S., Wong, A. & Kim, H.-J. Ovarian function suppression with luteinizing hormone-releasing hormone agonists for the treatment of hormone receptor-positive early breast cancer in premenopausal women. Front Oncol. 11, 700722 (2021).
Di Lauro, L. et al. Role of gonadotropin-releasing hormone analogues in metastatic male breast cancer: results from a pooled analysis. J. Hematol. Oncol. 8, 1–5 (2015).
Hofland, L. J. & Lamberts, S. W. Somatostatin receptors and disease: role of receptor subtypes. Baillieres Clin. Endocrinol. Metab. 10, 163–176 (1996).
Druker, B. J. et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Eng. J. Med 344, 1038–1042 (2001).
Gurung, A., Bhattacharjee, A., Ali, M. A., Al-Hemaid, F. & Lee, J. Binding of small molecules at interface of protein–protein complex–A newer approach to rational drug design. Saudi J. Biol. Sci. 24, 379–388 (2017).
Ran, X. & Gestwicki, J. E. Inhibitors of protein–protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 44, 75–86 (2018).
Mason, J. M., M. Müller, K. & Arndt, K. M. (2008). iPEP: peptides designed and selected for interfering with protein interaction and function. Biochem. Soc. Trans. 36, 1442–1447 (2008).
Bruno, B. J., Miller, G. D. & Lim, C. S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 4, 1443–1467 (2013).
Chen, X., Zaro, J. L. & Shen, W.-C. Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 65, 1357–1369 (2013).
Sikora, K. et al. c‐myc oncogene expression in colorectal cancer. Cancer 59, 1289–1295 (1987).
Little, C. D., Nau, M. M., Carney, D. N., Gazdar, A. F. & Minna, J. D. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 306, 194–196 (1983).
Blackwood, E. M. & Eisenman, R. N. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217 (1991).
Li, L., Sun, W., Zhang, Z. & Huang, Y. Time-staggered delivery of docetaxel and H1-S6A, F8A peptide for sequential dual-strike chemotherapy through tumor priming and nuclear targeting. J. Control Release 232, 62–74 (2016).
Bidwell, G. L. III et al. Thermally targeted delivery of a c-Myc inhibitory polypeptide inhibits tumor progression and extends survival in a rat glioma model. PloS one 8, e55104 (2013).
Soucek, L. et al. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 17, 2463–2472 (1998).
Soucek, L., Nasi, S. & Evan, G. Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ. 11, 1038–1045 (2004).
Soucek, L. et al. Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683 (2008).
Krumlauf, R. Hox genes in vertebrate development. Cell 78, 191–201 (1994).
Bhatlekar, S., Viswanathan, V., Fields, J. Z. & Boman, B. M. Overexpression of HOXA4 and HOXA9 genes promotes self‐renewal and contributes to colon cancer stem cell overpopulation. J. Cell Physiol. 233, 727–735 (2018).
Morgan, R. et al. Antagonism of HOX/PBX dimer formation blocks the in vivo proliferation of melanoma. Cancer Res. 67, 5806–5813 (2007).
Shen, W.-F. et al. Hox homeodomain proteins exhibit selective complex stabilities with Pbx and DNA. Nucleic Acids Res. 24, 898–906 (1996).
Neuteboom, S. & Murre, C. Pbx raises the DNA binding specificity but not the selectivity of antennapedia Hox proteins. Mol. Cell Biol. 17, 4696–4706 (1997).
Plowright, L., Harrington, K., Pandha, H. & Morgan, R. HOX transcription factors are potential therapeutic targets in non-small-cell lung cancer (targeting HOX genes in lung cancer). Br. J. Cancer 100, 470–475 (2009).
Moore, A. R., Rosenberg, S. C., McCormick, F. & Malek, S. RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Disco. 19, 533–552 (2020).
Huang, L., Guo, Z., Wang, F. & Fu, L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct. Target Ther. 6, 1–20 (2021).
Garrigou, M. et al. Accelerated identification of cell active KRAS inhibitory macrocyclic peptides using mixture libraries and automated ligand identification system (ALIS) technology. J. Med. Chem. 65, 8961–8974 (2022).
Lim, S. et al. Discovery of cell active macrocyclic peptides with on-target inhibition of KRAS signaling. Chem. Sci. J. 12, 15975–15987 (2021).
Lee, Y. et al. Targeted Degradation of Transcription Coactivator SRC‐1 through the N‐Degron Pathway. Angew. Chem. Int Ed. Engl. 59, 17548–17555 (2020).
Choi, S. R., Wang, H. M., Shin, M. H. & Lim, H.-S. Hydrophobic tagging-mediated degradation of transcription coactivator SRC-1. Int J. Mol. Sci. 22, 6407 (2021).
Acknowledgements
This work was supported by National Research Foundation grants [2021R1A5A2021614, 2021R1A2C1005513, and 2022M3A9G8095334] funded by the Ministry of Science and ICT of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vadevoo, S.M.P., Gurung, S., Lee, HS. et al. Peptides as multifunctional players in cancer therapy. Exp Mol Med 55, 1099–1109 (2023). https://doi.org/10.1038/s12276-023-01016-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01016-x
