O mal das montanhas e os neurônios do Monge
 A doença do Monge é uma condição fisiológica que atinge mais de 140 milhões de pessoas que estão expostas a grandes altitudes (mais que 2.500 metros do nível do mar) por tempos prolongados. Nos Andes, a prevalência chega a 20%, sugerindo que a maioria dos “highlanders” são saudáveis. Porém, os que sofrem com o mal da montanha crônico são afetados por uma série de condições neurológicas, como fatiga, dor de cabeça, confusão mental e perda de memória. O quadro pode ser fatal se agravar para um edema cerebral, por exemplo.
A doença do Monge é uma condição fisiológica que atinge mais de 140 milhões de pessoas que estão expostas a grandes altitudes (mais que 2.500 metros do nível do mar) por tempos prolongados. Nos Andes, a prevalência chega a 20%, sugerindo que a maioria dos “highlanders” são saudáveis. Porém, os que sofrem com o mal da montanha crônico são afetados por uma série de condições neurológicas, como fatiga, dor de cabeça, confusão mental e perda de memória. O quadro pode ser fatal se agravar para um edema cerebral, por exemplo.
É impossível prever quais indivíduos são mais susceptíveis a síndrome do Monge. Em geral, descobre-se apenas quando já está em altas altitudes. Também não há muito o que fazer, não existe um tratamento ideal ou cura para a condição. Para entender um pouco mais sobre as bases neuro-genéticas responsáveis pelos sintomas, nosso grupo colaborou com um outro laboratório, especializado na fisiologia humana em condições limitantes de oxigênio.
O grupo nos procurou interessado em reproduzir nosso trabalho com autismo, no modelo da doença do Monge. Ou seja, reprogramando células da pele de indivíduos afetados e saudáveis em neurônios no laboratório.
O primeiro desafio foi conseguir biopsia de pele dessa população andina. Foi necessário um trabalho de logística intenso, com coleta do material na região de Cerro de Pasco, no Peru, com uma elevação de 4.300 metros e transferência do tecido para San Diego, Califórnia. Tudo em tempo recorde, usando todo transporte possível, inclusive mulas. Assim que chegaram, as células da pele desses indivíduos foram reprogramadas para um estágio de pluripotência induzida, semelhante a de células-tronco embrionárias.
As células iPS foram então induzidas a se especializar em células do sistema nervoso, no caso neurônios excitatórios da região cortical. Em uma análise panorâmica morfológica, neurônios derivados dos pacientes eram muito semelhantes aos do grupo controle. Porém, do ponto de vista funcional, observamos uma alteração significativa.
Neurônios derivados dos pacientes eram menos excitáveis que os controle, ou seja, precisavam de mais tempo para processar e transmitir a informação elétrica. Na tentativa de desvendar as possíveis causas desse defeito funcional, descobrimos que os neurônios dos pacientes apresentavam quantidades inferiores de canais de sódio, importantes para o funcionamento neuronal.
Conforme previamente documentado por Carlos Monge ao descrever os sintomas em seus pacientes, o mal da montanha tem uma contribuição familiar e hereditária muito forte. É também mais frequente em homens europeus, comparado com outros grupos étnicos, sugerindo que o fator genético seja, de fato, relevante na fisiologia neural daqueles afetados pela condição.
Validamos alguns dos genes candidatos, mostrando que esses podem influenciar diretamente na regulação dos canais de sódio em neurônios humanos.
O trabalho acaba de ser publicado (Zhao e colegas, "Neuroscience" 2015) e pode levar os afetados a melhores tratamentos, usando-se drogas que atuem diretamente nos canais de sódio. Também promete ser uma ferramenta de diagnóstico interessante, auxiliando na seleção de profissionais que possam ser resistentes a altas altitudes, como atletas, por exemplo.
Foto: Martin St-Amant/Creative Commons

 Quem leu a biografia de Steve Jobs (fundador da Apple) deve ter notado que ele conseguiu estender sua batalha contra o letal câncer de pâncreas muito além do que a grande maioria dos pacientes com o mesmo tipo de câncer. Jobs conseguiu isso mesmo tendo tomado a decisão errada de recorrer a medicina alternativa logo após o diagnóstico. Uma das razões dessa sobrevida excepcional foi seu entusiasmo pela vida. A outra razão, acesso a tecnologia de ponta.
Quem leu a biografia de Steve Jobs (fundador da Apple) deve ter notado que ele conseguiu estender sua batalha contra o letal câncer de pâncreas muito além do que a grande maioria dos pacientes com o mesmo tipo de câncer. Jobs conseguiu isso mesmo tendo tomado a decisão errada de recorrer a medicina alternativa logo após o diagnóstico. Uma das razões dessa sobrevida excepcional foi seu entusiasmo pela vida. A outra razão, acesso a tecnologia de ponta. A história do uso da Cannabis na medicina é antiga. Mas talvez uma das situações mais relevantes que marcam a entrada da maconha no mercado farmacêutico foi a que aconteceu nos anos 90. Oficiais ingleses começaram a notar algo frequente acontecendo nos julgamentos de pessoas portando marijuana: um alto número de pacientes com esclerose múltipla justificavam o consumo alegando que a erva trazia relaxamento muscular e aliviava a dor. Em 1998, um comitê inglês de ciência e tecnologia encarregado de estudar o fenômeno, concluiu que a planta poderia dar origens a compostos de interesse médico.
A história do uso da Cannabis na medicina é antiga. Mas talvez uma das situações mais relevantes que marcam a entrada da maconha no mercado farmacêutico foi a que aconteceu nos anos 90. Oficiais ingleses começaram a notar algo frequente acontecendo nos julgamentos de pessoas portando marijuana: um alto número de pacientes com esclerose múltipla justificavam o consumo alegando que a erva trazia relaxamento muscular e aliviava a dor. Em 1998, um comitê inglês de ciência e tecnologia encarregado de estudar o fenômeno, concluiu que a planta poderia dar origens a compostos de interesse médico. O entendimento de doenças genéticas tem se beneficiado muito com os recentes avanços e progressos da genômica, especialmente no que se refere ao sequenciamento do DNA. A tecnologia tem evoluído tanto que em breve será possível sequenciar todos indivíduos do planeta, principalmente com a queda no custo operacional.
O entendimento de doenças genéticas tem se beneficiado muito com os recentes avanços e progressos da genômica, especialmente no que se refere ao sequenciamento do DNA. A tecnologia tem evoluído tanto que em breve será possível sequenciar todos indivíduos do planeta, principalmente com a queda no custo operacional.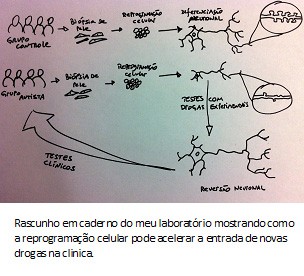 Em 2010, usamos uma
Em 2010, usamos uma 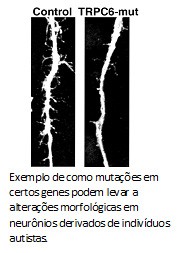 O que a Karina e o Allan mostraram foi que morfologia dos neurônios derivados desse autista mostrou-se menos complexa, com uma arborização menor do que o grupo controle. O número de sinapses e fisiologia das redes nervosas também estavam alteradas. Mas, ao contrário das formas sindrômicas do espectro, autistas clássicos possuem uma base genética complexa. Para entender o porquê dos defeitos nos neurônios desse paciente, decidimos sequenciar o genoma do paciente. Encontramos diversas mutações, inclusive uma que anulava uma das cópias do gene TRPC6. Esse gene codifica para um canal na membrana celular que permite a entrada de cálcio, um sinalizador para a formação de sinapses e maturação neuronal. Como o paciente possui apenas uma cópia funcional do gene TRPC6, isso explicaria a redução de sinapses e as alterações morfológicas observadas.
O que a Karina e o Allan mostraram foi que morfologia dos neurônios derivados desse autista mostrou-se menos complexa, com uma arborização menor do que o grupo controle. O número de sinapses e fisiologia das redes nervosas também estavam alteradas. Mas, ao contrário das formas sindrômicas do espectro, autistas clássicos possuem uma base genética complexa. Para entender o porquê dos defeitos nos neurônios desse paciente, decidimos sequenciar o genoma do paciente. Encontramos diversas mutações, inclusive uma que anulava uma das cópias do gene TRPC6. Esse gene codifica para um canal na membrana celular que permite a entrada de cálcio, um sinalizador para a formação de sinapses e maturação neuronal. Como o paciente possui apenas uma cópia funcional do gene TRPC6, isso explicaria a redução de sinapses e as alterações morfológicas observadas.