Mechanism
PI3K activation
There are multiple types of phosphoinositide 3-kinase but only class I are responsible for lipid phosphorylation in response to growth stimuli. Class 1 PI3Ks are heterodimers composed of a regulatory subunit p85 and a catalytic subunit p110, named by their molecular weights. [4]

The pathway can be activated by a range of signals, including hormones, growth factors and components of the extracellular matrix (ECM). [5] It is stimulated by binding of an extracellular ligand to a receptor tyrosine kinase (RTK) in the plasma membrane, causing receptor dimerization and cross-phosphorylation of tyrosine residues in the intracellular domains. The regulatory subunit p85 binds to phosphorylated tyrosine residues on the activated receptor via its Src homology 2 (SH2) domain. It then recruits the catalytic subunit p110 to form the fully active PI3K enzyme. Alternatively, adaptor molecule Grb2 binds to phospho-YXN motifs of the RTK and recruits p85 via Grb2-associated binding (GAB) scaffold protein. [6]
The p110 subunit can also be recruited independently of p85. For example, Grb2 can also bind the Ras-GEF Sos1, leading to activation of Ras. Ras-GTP then activates the p110 subunit of PI3K. Other adaptor molecules such as insulin receptor substrate (IRS) can also activate p110. [7]
PI3K can also be activated by G protein-coupled receptors (GPCR), via G-protein βγ dimers or Ras which bind PI3K directly. In addition, the Gα subunit activates Src-dependent integrin signaling which can activate PI3K. [8]
Phosphoinositide formation

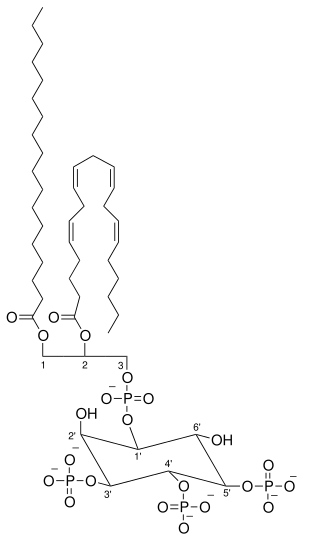
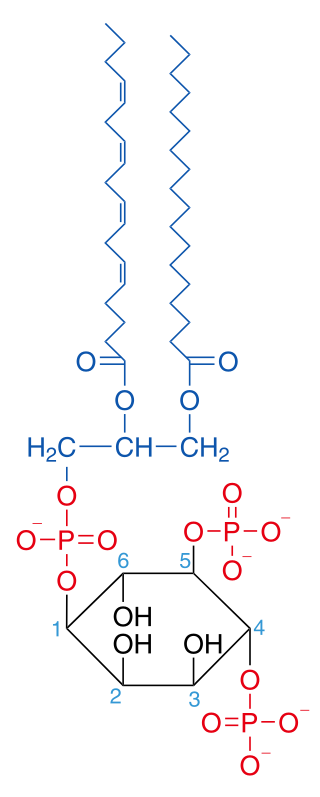
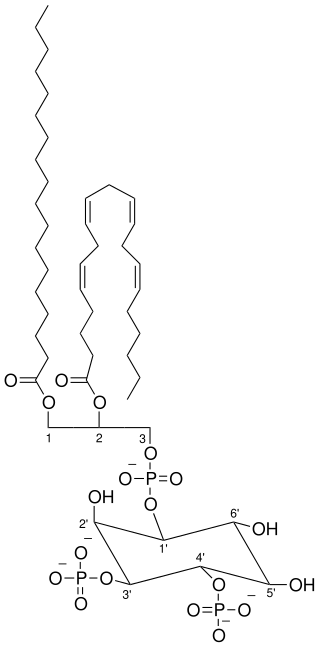
Activated PI3K catalyses the addition of phosphate groups to the 3'-OH position the inositol ring of phosphoinositides (PtdIns), producing three lipid products, PI(3)P, PI(3,4)P2 and PI(3,4,5)P3:
Phosphatidylinositol (PI) → PI 3-phosphate, (PI(4)P) → PI 3,4-bisphosphate, (PI(4,5)P2) → PI 3,4,5-triphosphate [9]
These phosphorylated lipids are anchored to the plasma membrane, where they can directly bind intracellular proteins containing a pleckstrin homology (PH) or FYVE domain. For example, the triphosphate form (PI(3,4,5)P3) binds Akt and phosphoinositide-dependent kinase 1 (PDK1) so they accumulate in close proximity at the membrane. [1] [10]
Akt activation
Akt resides in the cytosol in an inactive conformation, until the cell is stimulated and it translocates to the plasma membrane. The Akt PH domain has a high affinity for second messenger PI(3,4,5)P3, binding to it preferentially over other phosphoinositides. [11] Thus PI3K activity is essential for translocation of Akt to the membrane. Interaction with PI(3,4,5)P3 causes conformational changes and exposure of phosphorylation sites Thr308 in the kinase domain and Ser473 in the C-terminal domain. Akt is partially activated by phosphorylation of T308 by PDK1. Full activation requires phosphorylation of S473, which can be catalysed by multiple proteins, including phosphoinositide-dependent kinase 2 (PDK2), integrin-linked kinase (ILK), [1] mechanistic target of rapamycin complex complex 2 (mTORC2) and DNA-dependent protein kinase (DNA-PK). [12] [7] [13] The regulation of Ser473 phosphorylation is not fully understood but may also be influenced by autophosphorylation after Thr308 phosphorylation. After stimulation, the levels of PIP3 decrease and Akt activity is attenuated by dephosphorylation by serine/threonine phosphatases. [5]
PI3K-independent activation
Although PI3K is the major mode of Akt activation, other tyrosine or serine/threonine kinases have been shown to activate Akt directly, in response to growth factors, inflammation or DNA damage. These can function even when PI3K activity is inhibited. [14] Other studies have shown Akt can be activated in response to heat shock [15] or increases in cellular Ca2+ concentration, via Ca2+/Calmodulin-dependent protein kinase kinase (CAMKK). [13] [16]
| Activating Kinase | Akt Phosphorylation Site | Details |
|---|---|---|
| Activated CDC42 kinase 1 (Ack1) | Tyr176 | Akt binds preferentially to phosphatidic acid (PA) instead of PIP3 allowing translocation to the plasma membrane. [17] |
| Src | Tyr315, Tyr326 | Requires interaction of the Src SH3 domain and proline-rich region at the C-terminal of Akt. [18] |
| Protein tyrosine kinase 6 (PTK6) | Tyr215, Tyr315 and Tyr326 | Activates Akt in response to epidermal growth factor (EGF) [19] |
| IκB kinase ε (IKKε) | Ser137, Thr308 and Ser473 | Independent of the PH domain, PI3K, PDK1 and mTOR [20] |
| TANK-binding kinase 1 (TBK1) | Thr195, Ser378 and Ser473 | In response to Toll-like receptor activation in macrophages. [21] |
| DNA-dependent protein kinase (DNA-PK) | Ser473 | Activated by double-strand DNA breaks formed by ionizing radiation. [22] |