Синдром Елерса — Данлоса
Ця стаття містить правописні, лексичні, граматичні, стилістичні або інші мовні помилки, які треба виправити. (січень 2020) |
| Синдром Елерса-Данлоса | |
|---|---|
.png) Шкіра при Синдромі Елерса-Данлоса | |
| Спеціальність | медична генетика |
| Симптоми | skin hyperelasticityd[1] і joint hypermobilityd[2] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | LD28.1 |
| МКХ-10 | E75-E77 |
| DiseasesDB | 4131 |
| MeSH | D016464 |
| | |
Синдром Елерса-Данлоса (СЕД, англ. EDS) — група генетичних розладів сполучної тканини[3]. Симптоми можуть включати надмірні гнучкість кінцівок та еластичність шкіри, біль у суглобах, аномальне утворення рубців. Це може бути помічені при народженні або в ранньому дитинстві. Ускладнення можуть включати розшарування аорти, вивихи суглобів, сколіоз, хронічний біль або ранній остеоартрит.[4]
Синдром пов'язані з мутацією деяких з більш ніж десятка різних генів[3]. Специфічний вплив гена визначає специфічність синдрому. Деякі випадки є результатом нової мутації, яка відбувається під час раннього розвитку, в той час як інші успадковуються аутосомно-домінантним або рецесивним механізмом[3]. Це призводить до дефектів у структурі або в обробці колагену. Діагностика часто ґрунтується на симптомах і підтверджується генетичним тестуванням або біопсією шкіри. Пацієнтам часто встановлюють інші діагнози — іпохондрію, депресію або синдром хронічної втоми.
Лікування невідоме[5]. Фізична терапія і фіксація можуть сприяти зміцненню м'язів і суглобів. Ті види синдрому, які уражають кровоносні судини, зазвичай призводять до зменшення тривалості життя хворого.[5]
Синдроми уражають принаймні одного з 5000 осіб у всьому світі. Прогноз залежить від конкретного виду синдрому. Надмірну рухливість вперше описав Гіппократ у 400 р. До н. Е.[6] Синдроми названі на честь двох лікарів Едварда Елерса з Данії та Анрі-Александра Данлоса з Франції, які описали їх на рубежі XX-го століття[7].
Ознаки значно варіюються залежно від конкретного виду синдрому, який має людина. Ця група розладів впливає на сполучні тканини, частіше за все в суглобах, шкірі та кровоносних судинах, і викликає ефекти, починаючи від послаблених суглобів до ускладнень, що загрожують життю[8]. Основні ознаки та симптоми наведені нижче.
Симптоми опорно-рухового апарату включають гіпергнучкі суглоби, які є нестабільними і схильними до розтягування, дислокації, вивихів і гіперекстензії.[9][10]. Можуть спостерігатися ранній початок розвиненого остеоартриту,[11] хронічне дегенеративне захворювання суглобів[11], деформованість лебединої шиї на пальцях,[12] і деформація фаланг пальців. Може виникнути розрив сухожиль або м'язів[13]. Також можуть бути присутні деформації хребта, такі як сколіоз (викривлення хребта), кіфоз (грудний горб), прив'язаний синдром спинного мозку, і окципіатлантоаксиальна гіпермобільність[14]. Зустрічаються також міалгії (м'язові болі) і артралгії (болі в суглобах)[15], які можуть бути важкими і можуть призводити до інвалідності. Часто спостерігається ознака Тренделенбурга, що означає, що при стоянні на одній нозі таз падає з іншого боку[16]. Хвороба Осгуда-Шлаттера, хвороблива грудка на коліні, також поширена. У дітей грудного віку ходьба може бути відкладена (понад 18 місяців), а внизу — замість повзання[17].
-
 Людина з СЕД, що показує гіпермобільні пальці, включаючи мальформацію "лебедина шия" на 2-й 5-й фалангах, і гіпер-мобільні пальці
Людина з СЕД, що показує гіпермобільні пальці, включаючи мальформацію "лебедина шия" на 2-й 5-й фалангах, і гіпер-мобільні пальці -
 Людина з СЕД, що показує гіпер-мобільні пальці
Людина з СЕД, що показує гіпер-мобільні пальці -
 Людина з СЕД, що показує гіпер-мобільні метакарпофалангові суглоби
Людина з СЕД, що показує гіпер-мобільні метакарпофалангові суглоби



Слабка сполучна тканина є причиною вразливої шкіри, яка легко розривається[11], атрофічні рубці «сигаретного паперу» і легкі синці[18]. Виникають надлишкові шкірні складки, особливо на повіках[11][19]. Псевдопухлинні молюски[20], особливо на точках тиску. Рідше зустрічаються петехії[21], підшкірні сфероїди, [20] livedo reticularis, і п'єзогенні папули[22]. У судинної ЕЦП, тонка, напівпрозора і надзвичайно тендітна шкіра, яка легко розривається, також є симптомом.
-
 Атрофічний рубець у випадку СЕД
Атрофічний рубець у випадку СЕД -
 Напівпрозора шкіра при судинному СЕД
Напівпрозора шкіра при судинному СЕД -
 Людина з СЕД, що демонструє гіпереластичність шкіри
Людина з СЕД, що демонструє гіпереластичність шкіри

.png)

- Синдром торакального виходу[23]
- Артеріальний розрив[24]
- Захворювання клапанів серця, такі як пролапс мітрального клапана, створює підвищений ризик інфекційного ендокардиту під час операції. Це може перерости в небезпечну для життя ступінь. Аномалії провідності серця були виявлені у осіб з гіпермобільною формою СЕД[25].
- Дилатація та / або розрив (аневризма) висхідної аорти[26]
- Постуральний синдром ортостатичної тахікардії[27]
- Синдром Рейно
- Варикозне розширення вен[28]
- Шуми в серці
- Аномалії провідності серця

- Грижа Хіатала[20]
- Гастроезофагеальний рефлюкс[29]
- Порушення моторики шлунково-кишкового тракту[30]
- Дизаутономія[31]
- Знак Горліна (дотик язиком до носа)[32]
- Анальний пролапс[20]
- Спонтанний пневмоторакс[11]
- Нервові розлади (синдром зап'ястного каналу, акропарестезія, нейропатія, у тому числі нейропатія малого волокна)[33]
- Нечутливість до місцевих анестетиків.[34]
- Аномалія Арнольда — К'ярі[35]
- Неспроможність агрегації тромбоцитів (тромбоцити не злипаються належним чином)[36]
- Ускладнення вагітності: посилення болю, легка та помірна перипартулярная кровотеча, цервікальна недостатність, розривання матки або передчасний розрив мембран.
- Черепно-хребцева нестабільність: спричинена травмою ділянок голови і шиї, таких як струс мозку. Зв'язки в шиї нездатні заживати належним чином, тому структура шиї не має здатності підтримувати череп, який потім може потрапити в стовбур мозку, блокуючи нормальний потік спинномозкової рідини, що призводить до проблем, пов'язаних з відмовою вегетативної нервової системи правильно працювати.[37].
- Целіакія може бути пов'язана з СЕД. Крім того, вона може бути неправильно діагностується як EDS через загальні симптоми, включаючи втому, біль, шлунково-кишкові скарги, або серцево-судинну вегетативну дисфункцію[38].
Оскільки СЕД часто не діагностується або неправильно діагностується в дитинстві, деякі випадки були неправильно описані як жорстоке поводження з дітьми[39]. Біль, пов'язаний з розладами, може бути важким[40].

Тільки деякі випадки СЕД можуть бути позитивно ідентифіковані як прив'язані до конкретної генетичної варіації.
Мутації в цих генах можуть викликати СЕД:
- Волокнисті білки: COL1A1, COL1A2, COL3A1, COL5A1, COL5A2 і TNXB
- Ферменти: ADAMTS2, PLOD1, B4GALT7, DSE і D4ST1 / CHST14
Мутації в цих генах зазвичай змінюють структуру, створення або обробку колагену або білків, які взаємодіють з колагеном. Колаген забезпечує структуру і міцність сполучної тканини. Дефект колагену може послабити сполучну тканину в шкірі, кістках, кровоносних судинах і органах, що призводить до особливостей розладу. Схеми спадкування залежать від специфічного синдрому. Більшість форм СЕД успадковуються за аутосомно-домінантною схемою, що означає, що тільки одна з двох копій даного гена повинна бути змінена, щоб викликати розлад. Деякі з них успадковані в аутосомно-рецесивному порядку, що означає, що обидві копії гена повинні бути змінені для того, щоб людина була вражена порушенням. Вона також може бути індивідуальною (de novo або «спорадичною») мутацією[41].
Діагноз може бути підтверджений шляхом оцінки історії хвороби та клінічного спостереження. Критерії Beighton широко використовуються для оцінки ступеня гіпермобільності суглобів. ДНК і біохімічні дослідження можуть допомогти визначити уражених СЕД осіб. Діагностичні тести включають тестування колагенових генних мутацій, типування колагену через біопсію шкіри, ехокардіографію та виявлення активності лізилгидроксилази або оксидази. Однак ці тести не можуть підтвердити всі випадки, особливо у випадках немаптурованої мутації, тому клінічна оцінка генетиком залишається досить важливою. Якщо від СЕД страждають кілька осіб у сім'ї, то можливе проведення пренатальної діагностики за допомогою методу ДНК, відомого як дослідження зв'язків[42]. Знання про СЕД серед практикуючих лікарів є досить скудними[43][44]. Наразі проводяться дослідження з виявлення генетичних маркерів для всіх типів[45].
Станом на 2017 рік було охарактеризовано 13 СЕД, які мають суттєве перекриття характеристик[46].
Гіпермобільний СЕД (hEDS, раніше класифікований як тип 3) характеризується, в першу чергу, гіпермобільністю суглобів, що вражає як великі, так і малі суглоби, що може призвести до рецидивуючих вивихів суглобів та підвивихів (часткова дислокація суглобів). Загалом, люди з цим типом можуть мати м'яку, гладку і оксамитову шкіру з легкими синцями і хронічними болями в м'язах і/або кістках[46]. Мутація, що викликає цей тип СЕД, невідома. При цьому типі спостерігається менше ураження шкіри, ніж при інших типа. Генетичного тесту для цього типу немає.
Класичний СЕД (cEDS, раніше класифікований як тип 1) асоціюється з надзвичайно еластичною, гладкою та крихкою шкірою, що легко травмується; рубці після шрамів широкі та атрофічні; також наявна і гіпермобільність суглобів. Також часто зустрічаються псевдопухлинки моллюсоїдів (кальциновані гематоми над точками тиску, такими як лікті) і сфероїди (кісти, що містять жир на передпліччях і гомілках). Може виникнути гіпотонія і уповільнений руховий розвиток[46]. Мутація, що викликає цей тип СЕД, знаходиться в генах COL5A1, COL5A2 і COL1A1. Вона вражає шкіру більше, ніж hEDS.
Судинний СЕД (vEDS, раніше класифікований як тип 4) характеризується тонкою, напівпрозорою шкірою, яка надзвичайно тендітна і легко отримує синці. Артерії і деякі органи, такі як кишечник і матка, теж слабкі і схильні до розриву. Люди з цим типом зазвичай мають невеликий зріст і тонке волосся на голові. Вони також мають характерні риси обличчя, включаючи великі очі, низькоросле підборіддя, впалі щоки, тонкий ніс і губи, і вуха без мочок[47]. Присутня гіпермобільність суглобів, але в основному вона обмежена дрібними суглобами (пальцями рук та ніг). Інші загальні риси включають клишоногість, розрив сухожиль та/або м'язів, акрогерію (передчасне старіння шкіри рук і ніг), ранній початок варикозного розширення вен, пневмоторакс (колапс легені), рецесію ясен і зменшення кількості жиру під шкірою[46]. Цей тип може бути викликаний мутаціями гена COL3A1.

Кифоскаліозний СЕД (kEDS, раніше класифікований як тип 6) пов'язаний з важкою гіпотонією при народженні, затримкою розвитку моторних функцій, прогресуючим сколіозом (присутній від народження) і склеральною крихкістю. Люди з цим типом можуть також легко отримувати синці, можуть мати крихкі артерії, що схильні до розриву, незвично малі рогівки і остеопенію (низька щільність кісток). До інших поширених ознак можна віднести «марфаноїдний габітус», який характеризується довгими, стрункими пальцями (арахнодактилія), незвичайно довгими кінцівками і впалою грудною клітиною (pectus excavatum) або виступаючою грудною клітиною (pectus carinatum)[46]. Цей тип може бути викликаний мутаціями в гені PLOD1.
Артрохалазійний СЕД (aEDS, раніше класифікований як типи 7A & 7B) характеризується важкою гіпермобільністю суглобів і вродженим вивихом стегна. Інші поширені риси включають тендітну, еластичну шкіру, що легко травмується, гіпотонією, кіфосколіозом (кіфоз і сколіоз) і легкою остеопенією[46]. Зазвичай вражається колаген типу I. Цей тип дуже рідкісний, про що свідчать всього близько 30 зареєстрованих випадків. Цей тип проявляється більш тяжко, ніж гіпермобільний СЕД. Мутації в генах COL1A1 і COL1A2 викликають артрохалазійний СЕД.
Дерматоспараксійний СЕД (dEDS, раніше класифікований як тип 7C) пов'язаний з надзвичайно тендітною шкірою, що дуже просто, при цьому сильно травмується та рубцюється; людина з дерматоспараксійним СЕД має провисаючу, надлишкову шкіру, особливо на обличчі; і грижі. Це надзвичайно рідкісний тип, про що свідчать близько 10 зафіксований випадків.
Синдром крихкої рогівки характеризується тонкою роговою оболонкою, раннім початком прогресуючого кератоглобуса або кератоконуса, а також синьою склерою[46]. Часто зустрічаються класичні симптоми, такі як гіпермобільні суглоби та гіпереластична шкіра.
Класично подібний СЕД (clEDS) характеризується гіперекстенсією шкіри з оксамитовою текстурою шкіри та відсутністю атрофічного рубцювання, генералізованою гіпермобільностю суглобів з або без рецидивуючих вивихів (найчастіше плеча та гомілковостопного суглоба) та легко ушкоджуваною шкірою або спонтанним екхімозом (знебарвлення шкіри) внаслідок кровотечі під нею[46].
Спондилодіспластичний СЕД (spEDS) характеризується невисоким зростом (прогресуючим у дитинстві), м'язовою гіпотонією (від тяжкої вродженої, до легкої з пізнім початком) і викривленням кінцівок[46].
Мускульно-контрактуральний СЕД (mcEDS) характеризуються вродженими множинними контрактурами, характерними аддукційно-згинальними контрактурами та/або таліпами еквіноварусу (клишоногістю), характерними краніофациальними особливостями, які проявляються при народженні або в ранньому дитинстві, а також особливостями шкіри, такими як підвищена чутливість шкіри, синці, крихкість шкіри з атрофічними рубцями і підвищене долонне зморщування[46].
Міопатичний СЕД (mEDS) характеризується вродженою гіпотонією м'язів і/або атрофією м'язів, яка покращується з віком, контрактурами проксимальних суглобів (суглоби коліна, стегна і ліктя) і гіпермобільністю дистальних суглобів (суглобів щиколоток, зап'ясток, ніг і рук)[46].
Пародонтальний СЕД (pEDS) характеризується важким і непереборним періодонтитом з дитинства або підліткового віку, недостатністю прикріплення ясен, предбіальними бляшками і сімейною історією хвороби родича першого ступеня, що відповідає клінічним критеріям синдрому[46].
Серцево-клапанний СЕД (cvEDS) характеризується вираженими прогресуючими серцево-клапанними проблемами (аортальний клапан, мітральний клапан), проблемами зі шкірою (гіперекстенівність, атрофічні рубці, тонка шкіра, що легко травмується) і гіпермобільністю суглобів (генералізована або обмежена дрібними суглобами)[46].
Деякі розлади мають схожі характеристики з СЕД. Наприклад, при cutis laxa, шкіри у хворої людини забагато, вона провисає та морщиться. При СЕД шкіра може бути відтягнута від тіла, але вона еластична і повертається до нормального стану, коли відпускається. При синдромі Марфана суглоби дуже рухливі і стаються подібні до СЕД серцево-судинні ускладнення. Люди з СЕД мають тенденцію мати марфаноїдний зовнішній вигляд (наприклад, худі, довгі руки і ноги, «павукові» пальці). Однак, зовнішній вигляд і особливості в деяких випадках СЕД також мають диференціюючі характеристики, включаючи короткий зріст, великі очі, а також маленький рот і підборіддя, обумовлені невеликим піднебінням. Піднебіння може мати високу дугу, що викликає стоматологічне витіснення. Кровоносні судини іноді можна легко побачити через напівпрозору шкіру, особливо на грудях. Генетичний розлад сполучної тканини, синдром Лойса-Дітца, також має симптоми, які перекриваються з СЕД.
У минулому хвороба Менкеса, розлад метаболізму міді, вважалася СЕД. Людям нерідко невірно ставлять діагнози фіброміалгія, порушення згортання крові або інші порушення, які можуть імітувати симптоми СЕД. Через такі подібні розлади і ускладнення, що можуть виникнути в результаті недіагностованого випадку СЕД, важливо правильний діагностувати хвороби[49]. Pseudoxanthoma elasticum (PXE) варто враховувати при діагностиці[50].
До 1997 року система класифікації СЕД включала 10 конкретних типів, а також визнавала існування інших надзвичайно рідкісних типів. Пізніше система класифікації зазнала капітального ремонту і була зведена до шести основних типів з використанням описових назв. Генетичні фахівці визнають, що існують інші типи цього стану, але вони були задокументовані лише в одиночних випадках. За винятком гіпермобільності (тип 3), найбільш поширеного типу з усіх десяти, деякі з специфічних мутацій були ідентифіковані, і вони можуть бути точно визначені за допомогою генетичного тестування; це дуже важливо через велике значення варіабельності в окремих випадках. Однак, негативні результати генетичних тестів не виключають діагнозу, оскільки не всі мутації виявлені; тому клінічні прояви є дуже важливими[51].
Форми СЕД в цій категорії можуть характеризуватись м'якою, слабо-розтягуваною шкірою, укороченими кістками, хронічною діареєю, гіпермобільністю суглобів і вивихами, розривами сечового міхура або поганим загоєнням ран. Схеми успадкування в цій групі включають рецесивну, аутосомно-домінантну і аутосомно-рецесивну. Приклади типів споріднених синдромів, відмінних від вищезазначених у медичній літературі, включають[52]:
- 305200 [Архівовано 13 березня 2020 у Wayback Machine.] — тип 5
- [1] [Архівовано 23 червня 2019 у Wayback Machine.] — тип 8 — невизначений ген, локус 12p13
- 225310 [Архівовано 13 березня 2020 у Wayback Machine.] — тип 10 — невизначений ген, локус 2q34
- 608763 [Архівовано 11 липня 2020 у Wayback Machine.] — тип Beasley–Cohen
- 130070 [Архівовано 9 червня 2019 у Wayback Machine.] — прогероідна форма — B4GALT7
- 130090 [Архівовано 10 липня 2020 у Wayback Machine.] — невизначений тип
- 601776 [Архівовано 23 червня 2019 у Wayback Machine.] — D4ST1-дефіцитний синдром Елерса-Данлоса (синдром прищепленої пальцевої клинці) CHST14
На 2018 рік ефективного лікування синдромів Елерса-Данлоса не розроблено; воно зводиться до підтримки життя. Можуть бути корисними ретельний моніторинг серцево-судинної системи, фізіотерапія, трудова терапія та ортопедичні інструменти (наприклад, інвалідні коляски, кріплення, зовнішні скелети). Це може допомогти у стабілізації суглобів і запобіганні травм. Ортопедичні інструменти допомагають запобігти подальшому пошкодженню суглобів, особливо довгостроково, хоча індивідуумам рекомендується не починати залежати від них, поки інші варіанти мобільності не вичерпані. Люди повинні уникати дій, які призводять до блокування або надмірного розгинання суглобів[53].
Лікар може призначити гіпсування для стабілізації суглобів. Лікарі можуть направити особу на ортопедичне лікування. Також можуть призначатись консультації з фізіотерапевтом, щоб допомогти зміцнити м'язи та навчити людей правильно використовувати та зберігати свої суглоби[54][55].
Водна терапія сприяє розвитку і координації м'язів[56]. При мануальній терапії суглоб обережно мобілізується в межах руху та / або маніпуляцій[54][55]. Якщо консервативна терапія не допомагає, може знадобитися хірургічне втручання в суглоби. Можуть бути призначені препарати для зменшення болю або для лікування серцевих, травних або інших пов'язаних з ними станів. Для зменшення синців і поліпшення загоєння ран деякі пацієнти реагують на вітамін С[57]. Спеціальні запобіжні заходи часто мають проводитись медичними працівниками через велику кількість ускладнень, які, як правило, виникають у людей з СЕД. У судинних СЕД ознаками грудної або черевної болі вважаються травматичні ситуації[58].
Канабіноїди та медична марихуана показали певну ефективність у зниженні рівня болю[59].
Загалом, медичне втручання обмежується симптоматичною терапією. Перед вагітністю люди, які мають СЕД, повинні пройти генетичне консультування і ознайомитися з ризиками для власного тіла, які створює вагітність. Діти з СЕД повинні отримувати інформацію про свій стан, щоб вони могли зрозуміти, чому повинні уникати контактних видів спорту та інших фізично стресових дій. Дітей слід навчити, що демонстрація незвичайних положень кінцівок, які вони можуть підтримувати через вільні суглоби, не повинна проводитися задля втіхи, оскільки це може призвести до ранньої дегенерації суглобів. Емоційна підтримка поряд з поведінковою та психологічною терапією може бути корисною. Групи підтримки можуть бути надзвичайно корисними для людей, які мають справу з основними змінами в житті та поганим здоров'ям у зв'язку з СЕД. Члени сім'ї, вчителі та друзі повинні бути поінформовані про СЕД, щоб вони могли допомагати дитині[60].
Нестабільність суглобів, що призводить до виснаження і болю в суглобах, часто вимагає хірургічного втручання у людей з СЕД. Нестабільність майже всіх суглобів може статися, але найчастіше такі стани з'являються в нижніх і верхніх кінцівках, при цьому найбільш поширені зони — зап'ястя, пальці, плечі, коліна, стегна і щиколотки[54].
Загальними хірургічними процедурами є обробка суглобів, заміна сухожиль, капсулографія та артропластика. Після операції ступінь стабілізації, зменшення болю і задоволеність людей можуть покращитися, але хірургічне втручання не гарантує оптимального результату: уражені люди і хірурги повідомляють про часту незадоволеність результатами. Консенсус полягає в тому, що консервативне лікування є більш ефективним, ніж хірургічне[25], особливо тому, що у людей є додаткові ризики хірургічних ускладнень внаслідок захворювання. Три основні хірургічні проблеми, що виникають через СЕД:
- сила тканин зменшується, що робить тканину менш придатною для операції;
- крихкість кровоносних судин може викликати проблеми під час операції;
- загоєння рани часто затримується або неповне[54].
Якщо розглядати хірургічне втручання, доцільно звертатися за допомогою до хірурга з великими знаннями та досвідом у лікуванні людей, які страждають на СЕД та спільні проблеми з гіпермобільністю.
Місцеві анестетики, артеріальні катетери та центральні венозні катетери викликають підвищений ризик утворення синяків у людей з СЕД[61]. Люди з СЕД також виявляють стійкість до місцевих анестетиків. Резистентність до ксилокаїну і бупівакаїну не є рідкістю, і карбокаїн має тенденцію працювати краще у людей з СЕД. Спеціальні рекомендації по анестезії у людей з СЕД готуються за допомогою орфанної анестезії та розглядають всі аспекти анестезії для людей з ЕЦП[62]. Для підвищення безпеки слід використовувати детальні рекомендації щодо анестезії та періопераційної допомоги людям з СЕД[63].
Операції у людей з СЕД вимагають ретельної обробки тканин і більш тривалої іммобілізації після цього.
Прогноз для пацієнтів залежить від конкретного типу СЕД, який вони мають. Симптоми варіюють по тяжкості, навіть у тому ж самому розладі, і частота ускладнень змінюється. Деякі люди мають незначні симптоми, інші ж сильно обмежені в повсякденному житті. Екстремальна нестабільність суглобів, хронічний кістковий біль, дегенеративне захворювання суглобів, часті травми і деформації хребта можуть обмежувати рухливість. Важкі спінальні деформації можуть впливати на дихання. У випадку надзвичайної нестабільності суглобів, дислокації можуть виникати внаслідок простих завдань, таких як поворот в ліжку або поворот дверної ручки. Вторинні стани, такі як вегетативна дисфункція або серцево-судинні проблеми, що виникають у будь-якому типі, можуть впливати на прогноз і якість життя. Важка інвалідність, пов'язана з мобільністю, частіше спостерігається у гіпермобільних СЕД, ніж у класичних СЕД або судинних СЕД.
Хоча всі типи СЕД потенційно небезпечні для життя, більшість людей мають нормальну тривалість життя. Однак у тих, у кого є крихкість кровоносних судин, існує високий ризик ускладнень зі смертельними наслідками, включаючи спонтанний артеріальний розрив, який є найбільш поширеною причиною раптової смерті. Середня тривалість життя в популяції з судинними СЕД становить 48 років[64].
Синдроми Елерса-Данлоса є успадкованими розладами, які, за оцінками, зустрічаються приблизно в одному з 5000 новонароджених у всьому світі. Спочатку оцінки поширеності становили від 250 000 до 500 000 осіб, але ці оцінки незабаром виявилися занадто низькими, оскільки розлади отримали подальше дослідження, а медичні працівники стали більш вмілими у діагностиці. СЕД можуть бути набагато більш поширеними, ніж прийняті в даний час оцінки, зважаючи на широкий діапазон тяжкості, який притаманний розладам[65].
Поширеність розладів різко відрізняється. Найбільш часто зустрічається гіпермобільний СЕД, за якою слідує класичний СЕД. Інші дуже рідкісні. Наприклад, в усьому світі було описано менше 10 немовлят і дітей з дерматоспараксією. Деякі СЕД частіше зустрічаються в ашкеназських євреїв. Наприклад, шанс бути носієм СЕД дерматоспараксису є одним з 248 у євреїв ашкеназі, тоді як поширеність цієї мутації в загальній популяції становить один з 2000[66].

У 19 столітті кілька артистів цирків були представлені в якості «Еластичного чоловіка», «Індійського гумового чоловіка» і «Жабиного хлопчика». Список включав таких відомих людей (у свій час), як Фелікс Верль, Джеймс Морріс і Ейвері Чайлдс.[67]
Кілька сучасних знаменитостей мають СЕД
- Актриса Шеріл Хьюстон має гіпермобільний СЕД і використовує інвалідний візок[68]
- СЕД, можливо, зробив свій внесок у майстерність віртуозного скрипаля Ніколо Паганіні, оскільки він міг охоплювати пальцями більше, ніж звичайний скрипаль.[69]
- Кінозірка фільмів для дорослих Манді Морбід обговорює вплив її СЕД на рухливість і її життя.[70]
- Rei Haycraft, співак хард-рок групи Raimee, художник і ілюстратор, створив пісні і документальний фільм про життя з СЕД, а також написав пісні про її вплив на життя.[71][72]
- Американська активістка з прав на непрацездатних Анні Сегарра має СЕД і розповідає про свій стан на каналі в YouTube.[73]
- Гарі «Стретч» Тернер (показаний праворуч), артист цирку з Circus Of Horrors, має СЕД. Він володіє поточним рекордом Гіннеса з найбільш еластичної шкіри — 6 дюймів.
- Актриса / ведуча Jameela Jamil публічно розкрила свій діагноз синдрому Елерса-Данлоса[74].
- Учасник 11 сезону шоу «Королівські перегони Ру Пола» — Yvie Oddly у одному з випусків шоу розказав про те, що живе за СЕД[75]
Виявлено, що синдроми, подібні СЕД, є спадковими у гімалайських кішок, деяких домашніх короткошерстих кішок[76] і у деяких пород великої рогатої худоби[77]. Він розглядається як спорадичний стан у домашніх собак.
- Animal EDS
-
 СЕД у собаки
СЕД у собаки -
 СЕД у тієї ж собаки
СЕД у тієї ж собаки -
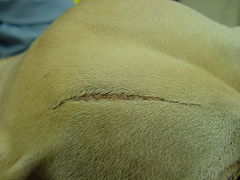 СЕД в тієї ж собаки, демонстрація атрофічного рубця
СЕД в тієї ж собаки, демонстрація атрофічного рубця -
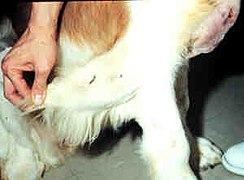 Сенбернар з СЕД демонструє надлишкову шкіру
Сенбернар з СЕД демонструє надлишкову шкіру




Дегенеративний десміт суспензіонної зв'язки (DSLD) є подібним станом, що спостерігається у багатьох порід коней. Спочатку він був визначений в перуанському Пасо і вважався умовою перевтоми і старшого віку. Однак, DSLD визнається у всіх вікових групах і на всіх рівнях діяльності. Це спостерігалося навіть у новонароджених лошат. Останнє дослідження призвело до перейменування хвороби як системного накопичення протеоглікану кінцівок, після того, як можливі системні та спадкові компоненти були окреслені Університетом Джорджії[78][79].
- ↑ https://www.ehlers-danlos.org/information/the-skin-in-hypermobile-ehlers-danlos-syndrome/
- ↑ https://www.nhs.uk/conditions/ehlers-danlos-syndromes/
- ↑ а б в Ehlers–Danlos syndrome". Genetics Home Reference. Archived from the original on 8 May 2016. Retrieved 8 May 2016
- ↑ Anderson, Bryan E. (2012). The Netter Collection of Medical Illustrations - Integumentary System E-Book (англ.) (вид. 2). Elsevier Health Sciences. с. 235. ISBN 978-1455726646. Архів оригіналу за 5 листопада 2017.
{{cite book}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ а б Ferri, Fred F. (2016). Ferri's Netter Patient Advisor (англ.). Elsevier Health Sciences. с. 939. ISBN 9780323393249. Архів оригіналу за 5 листопада 2017.
{{cite book}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Beighton, Peter H.; Grahame, Rodney; Bird, Howard A. (2011). Hypermobility of Joints. Springer. p. 1. ISBN 9781848820852. Archived from the original on 2017-11-05.
- ↑ Byers PH, Murray ML (2012). «Heritable collagen disorders: the paradigm of Ehlers–Danlos syndrome». Journal of Investigative Dermatology. 132 (E1): E6–11. doi:10.1038/skinbio.2012.3. PMID 23154631.
- ↑ Ehlers-Danlos syndrome. Genetic Home Reference. Архів оригіналу за 8 травня 2016. Процитовано 4 квітня 2018.
- ↑ Byers PH, Murray ML (2012). «Heritable collagen disorders: the paradigm of Ehlers–Danlos syndrome». Journal of Investigative Dermatology. 132 (E1): E6–11.
- ↑ Lawrence EJ (2005). The clinical presentation of Ehlers–Danlos syndrome. Adv Neonatal Care. 5 (6): 301—14. doi:10.1016/j.adnc.2005.09.006. PMID 16338669.
- ↑ а б в г д Ehlers–Danlos Syndrome. Mayo Clinic. Архів оригіналу за 25 June 2012. Процитовано 25 травня 2012.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Erçöçen AR, Yenidünya MO, Yilmaz S, Ozbek MR (1997). Dynamic swan neck deformity in a patient with Ehlers–Danlos syndrome. J Hand Surg Br Vol. 22 (1): 128—30. doi:10.1016/S0266-7681(97)80039-3. PMID 9061548.
- ↑ Vascular Type-EDS. Ehlers–Danlos Syndrome Network C.A.R.E.S. Inc. Архів оригіналу за 4 червня 2012. Процитовано 15 квітня 2019.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA (December 2007). Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. Journal of Neurosurgery. Spine. 7 (6): 601—9. doi:10.3171/SPI-07/12/601. PMID 18074684.
- ↑ Gedalia A, Press J, Klein M, Buskila D (1993). Joint hypermobility and fibromyalgia in schoolchildren. Annals of the Rheumatic Diseases. 52 (7): 494—6. doi:10.1136/ard.52.7.494. PMC 1005086. PMID 8346976.
- ↑ Dommerholt, Jan (27 січня 2012). CSF Ehlers Danlos Colloquium, Dr Jan Dommerholt. Chiari & Syringomyelia Foundation. Архів оригіналу за 4 May 2013. Процитовано 10 червня 2013.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Ehlers-Danlos syndrome - Diagnosis - Approach. BMJ Best Practice. 13 грудня 2016. Архів оригіналу за 19 August 2010. Процитовано 18 серпня 2017.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Cheong, J E L. (1 липня 2005). Unusual scars in a young female patient. Postgraduate Medical Journal. 81 (957): e4—5. doi:10.1136/pgmj.2004.026419. ISSN 1469-0756. PMC 1743308. PMID 15998813. Архів оригіналу за 18 січня 2017.
- ↑ Ehlers Danlos Syndrome - Morphopedics. morphopedics.wikidot.com (англ.). Архів оригіналу за 15 червня 2018. Процитовано 15 червня 2018.
- ↑ а б в Classical Type-EDS. Ehlers–Danlos Syndrome Network C.A.R.E.S Inc. Архів оригіналу за 30 травня 2012. Процитовано 15 квітня 2019.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Wilkins, Lippincott Williams & (2007). Portable Signs and Symptoms (англ.). Lippincott Williams & Wilkins. с. 465. ISBN 9781582556796. Архів оригіналу за 5 листопада 2017.
{{cite book}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Piezogenic papules - DermNet New Zealand. www.dermnetnz.org. Архів оригіналу за 26 листопада 2016.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Ericson Jr, William B; Wolman, Roger (March 2017). Orthopaedic management of the Ehlers–Danlos syndromes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 175 (1): 188—194. doi:10.1002/ajmg.c.31551. PMID 28192621.
- ↑ Byers PH, Murray ML (2012). Heritable collagen disorders: the paradigm of Ehlers–Danlos syndrome. Journal of Investigative Dermatology. 132 (E1): E6—11. doi:10.1038/skinbio.2012.3. PMID 23154631.
- ↑ а б Camerota F, Castori M, Celletti C, Colotto M, Amato S, Colella A, Curione M, Danese C (2014). Heart rate, conduction and ultrasound abnormalities in adults with joint hypermobility syndrome/Ehlers–Danlos syndrome, hypermobility type. Clin. Rheumatol. 33 (7): 981—7. doi:10.1007/s10067-014-2618-y. PMID 24752348.
- ↑ Wenstrup, R.J та ін. (2001). The Ehlers–Danlos Syndromes: Management of Genetic Syndromes. с. 131—149.
- ↑ Grigoriou, Emmanouil; Boris, Jeffrey R.; Dormans, John P. (February 2015). Postural Orthostatic Tachycardia Syndrome (POTS): Association with Ehlers-Danlos Syndrome and Orthopaedic Considerations. Clinical Orthopaedics and Related Research. 473 (2): 722—728. doi:10.1007/s11999-014-3898-x. PMC 4294907. PMID 25156902.
- ↑ Raffetto, JD; Khalil, RA (April 2008). Mechanisms of varicose vein formation: valve dysfunction and wall dilation. Phlebology. 23 (2): 85—98. doi:10.1258/phleb.2007.007027. PMID 18453484.
- ↑ Zeitoun, Jean-David; Lefèvre, Jérémie H.; de Parades, Vincent; Séjourné, César та ін. (November 2013). Functional Digestive Symptoms and Quality of Life in Patients with Ehlers-Danlos Syndromes: Results of a National Cohort Study on 134 Patients. PLoS One. 8 (11): e80321. Bibcode:2013PLoSO...880321Z. doi:10.1371/journal.pone.0080321. PMC 3838387. PMID 24278273.
{{cite journal}}: Обслуговування CS1: Сторінки із непозначеним DOI з безкоштовним доступом (посилання) - ↑ Brockway, Laura (April 2016). Gastrointestinal manifestations of Ehlers–Danlos syndrome (hypermobility type). Ehlers–Danlos Support UK. Архів оригіналу за 14 листопада 2016.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Ehlers–Danlos Syndrome. Underlying Causes of Dysautonomia. Dysautonomia International. 2012. Архів оригіналу за 18 грудня 2014.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Létourneau Y, Pérusse R, Buithieu H (2001). Oral manifestations of Ehlers–Danlos syndrome. J Can Dent Assoc. 67 (6): 330—4. PMID 11450296. Архів оригіналу за 15 грудня 2016.
{{cite journal}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Ehlers–Danlos syndrome: Definition from. Answers.com. Архів оригіналу за 6 березня 2014. Процитовано 27 лютого 2014.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Arendt-Nielsen, Lars. Patients Suffering from Ehlers Danlos Syndrome Type III Do Not Respond to Local Anesthetics. Архів оригіналу за 5 квітня 2015.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Castori M, Voermans NC (2014). Neurological manifestations of Ehlers-Danlos syndrome(s): A review. Iran J Neurol. 13 (4): 190—208. PMC 4300794. PMID 25632331.
{{cite journal}}: Cite має пустий невідомий параметр:|df=(довідка)Обслуговування CS1: Сторінки з параметром url-status, але без параметра archive-url (посилання) - ↑ MedlinePlus Encyclopedia Ehlers-Danlos syndrome
- ↑ Henderson, Fraser (2015). Indices of Cranio-vertebral Instability. Funded Research. Chiari & Syringomyelia Foundation. Архів оригіналу за 16 вересня 2016.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Levy, Howard P. (21 червня 2016). Hypermobile Ehlers-Danlos Syndrome. Архівована копія (Review). Seattle, WA: University of Washington, Seattle. PMID 20301456. Архів оригіналу за 18 січня 2017. Процитовано 15 квітня 2019.
{{cite book}}: Обслуговування CS1: Сторінки з текстом «archived copy» як значення параметру title (посилання) - ↑ Santschi, Darrell R. (3 квітня 2008). Redlands mother stung by untrue suspicions presses for accountability in child abuse inquiries. The Press Enterprise. Архів оригіналу за 28 лютого 2009.
- ↑ Chopra, Pradeep; Tinkle, Brad; Hamonet, Claude; Brock, Isabelle; Gompel, Anne; Bulbena, Antonio; Francomano, Clair (2017). Pain management in the Ehlers–Danlos syndromes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics (англ.). 175 (1): 212—219. doi:10.1002/ajmg.c.31554. ISSN 1552-4876. PMID 28186390.
{{cite journal}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ EDS Types | The Ehlers Danlos Society. The Ehlers Danlos Society (амер.). Архів оригіналу за 24 червня 2017. Процитовано 22 травня 2017.
{{cite news}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Sobey, Glenda (1 січня 2015). Синдром Елера-Данлоса: як діагностувати і коли проводити генетичні тести. Archives of Disease in Childhood (англ.). 100 (1): 57—61. doi:10.1136/archdischild-2013-304822. ISSN 0003-9888. PMID 24994860. Архів оригіналу за 28 травня 2020. Процитовано 27 квітня 2019.
- ↑ Ross, J.; Grahame, R. (2011). Joint hypermobility syndrome. BMJ. 342: c7167. doi:10.1136/bmj.c7167. PMID 21252103.
- ↑ Castori, Marco (2012). Ehlers–Danlos Syndrome, Hypermobility Type: An Underdiagnosed Hereditary Connective Tissue Disorder with Mucocutaneous, Articular, and Systemic Manifestations. ISRN Dermatology. 2012: 1—22. doi:10.5402/2012/751768. PMC 3512326. PMID 23227356.
{{cite journal}}: Обслуговування CS1: Сторінки із непозначеним DOI з безкоштовним доступом (посилання) - ↑ Типи СЕД. Асоціація Енлера-Данлоса (амер.). Архів оригіналу за 21 квітня 2020. Процитовано 17 жовтня 2018.
- ↑ а б в г д е ж и к л м н п Ehlers-Danlos syndromes. rarediseases.info.nih.gov (англ.). 20 квітня 2017. Архів оригіналу за 24 September 2017. Процитовано 23 вересня 2017.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Inokuchi, Ryota; Kurata, Hideaki; Endo, Kiyoshi; Kitsuta, Yoichi; Nakajima, Susumu; Hatamochi, Atsushi; Yahagi, Naoki (2 грудня 2014). Vascular Ehlers-Danlos Syndrome Without the Characteristic Facial Features. Medicine. 93 (28): e291. doi:10.1097/MD.0000000000000291. ISSN 0025-7974. PMC 4603083. PMID 25526469.
- ↑ Germain, DP. Ehlers-Danlos syndrome type IV. „Orphanet Journal of Rare Diseases”. 2. 32, 2007. PMID 17640391.
- ↑ Ehlers–Danlos Syndrome. Rarediseases.about.com. 25 травня 2006. Архів оригіналу за 12 квітня 2014. Процитовано 27 лютого 2014.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Reference, Genetics Home. Pseudoxanthoma elasticum. Genetics Home Reference (англ.). Архів оригіналу за 18 квітня 2018. Процитовано 17 квітня 2018.
- ↑ Що таке СЕД | The Ehlers–Danlos National Foundation. www.ednf.org. Архів оригіналу за 26 квітня 2016. Процитовано 6 січня 2016.
- ↑ OMIM Entry Search - ehlers-danlos syndrome. www.omim.org. Архів оригіналу за 8 липня 2020. Процитовано 27 квітня 2019.
- ↑ Physiotherapy and self-management – The Ehlers-Danlos Support UK. www.ehlers-danlos.org (англ.). Архів оригіналу за 8 грудня 2019. Процитовано 17 квітня 2018.
- ↑ а б в г Rombaut L, Malfait F, De Wandele I, Cools A, Thijs Y, De Paepe A, Calders P (July 2011). Medication, Surgery, and Physiotherapy Among Patients With the Hypermobility Type of Ehlers–Danlos Syndrome. Archives of Physical Medicine and Rehabilitation. 92 (7): 1106—12. doi:10.1016/j.apmr.2011.01.016. PMID 21636074.
- ↑ а б Woerdeman LA, Ritt MJ, Meijer B, Maas M (2000). Wrist problems in patients with Ehlers–Danlos syndrome. European Journal of Plastic Surgery. 23 (4): 208—210. doi:10.1007/s002380050252.
- ↑ Callewaert B, Malfait F, Loeys B, De Paepe A (March 2008). Ehlers–Danlos syndromes and Marfan syndrome. Best Practice & Research. Clinical Rheumatology. 22 (1): 165—189. doi:10.1016/j.berh.2007.12.005. PMID 18328988.
- ↑ Genetics of Ehlers–Danlos Syndrome~treatment на eMedicine
- ↑ Vascular (VEDS) Emergency Information | The Ehlers Danlos Society. The Ehlers Danlos Society (амер.). Архів оригіналу за 18 квітня 2018. Процитовано 17 квітня 2018.
- ↑ Topics in Pain Management: pain management in patients with hyper mobility disorders: frequently missed causes of chronic pain (PDF). Topics in Pain Management. Архів оригіналу (PDF) за 25 червня 2021. Процитовано 29 квітня 2019.
- ↑ M., Giroux, Catherine M.|Corkett, Julie K.|Carter, Lorraine (2016). The Academic and Psychosocial Impacts of Ehlers-Danlos Syndrome on Postsecondary Students: An Integrative Review of the Literature. Journal of Postsecondary Education and Disability (англ.). 29 (4): 414. Архів оригіналу за 18 квітня 2018. Процитовано 29 квітня 2019.
- ↑ Parapia, Liakat A.; Jackson, Carolyn (2008). Ehlers–Danlos syndrome – a historical review. British Journal of Haematology. 141 (1): 32—5. doi:10.1111/j.1365-2141.2008.06994.x. PMID 18324963.
- ↑ OrphanAnesthesia Guidelines
- ↑ Wiesmann, Thomas; Castori, Marco; Malfait, Fransiska; Wulf, Hinnerk (2014). Recommendations for anesthesia and perioperative management in patients with Ehlers–Danlos syndrome(s). Orphanet Journal of Rare Diseases. 9: 109. doi:10.1186/s13023-014-0109-5. PMC 4223622. PMID 25053156.
{{cite journal}}: Cite має пустий невідомий параметр:|df=(довідка)Обслуговування CS1: Сторінки із непозначеним DOI з безкоштовним доступом (посилання) - ↑ Pepin, Melanie; Schwarze, Ulrike; Superti-Furga, Andrea; Byers, Peter H. (2000). Clinical and Genetic Features of Ehlers–Danlos Syndrome Type IV, the Vascular Type. New England Journal of Medicine. 342 (10): 673—80. doi:10.1056/NEJM200003093421001. PMID 10706896.
- ↑ Ehlers–Danlos Syndrome: Epidemiology. Medscape.com. Архів оригіналу за 24 квітня 2013. Процитовано 27 лютого 2014.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Ehlers-Danlos syndrome type VIIc. geneaware.clinical.bcm.edu (амер.). Архів оригіналу за 14 серпня 2017. Процитовано 24 липня 2017.
{{cite web}}: Cite має пустий невідомий параметр:|df=(довідка) - ↑ Mitchell, Michael (1979). Монстри позолоченого віку: фотографії Charles Eisenmann. Торонто: Gage. с. 99, 100. ISBN 978-0771595219. OCLC 6892718.
- ↑ /a221287/houston-hits-out-at-preconceived-ideas.html Х'юстон показується на "попередні ідеї" - Новини коронації вулиць - мила. Digital Spy. 22 травня 2010. Архів оригіналу за 9 травня 2013. Процитовано 2014 - 02-27.
- ↑ Yücel D (1995). Чи Паганіні народився з фенотипом 4 або 3 синдрому Елерса-Данлоса?. Clin. Chem. 41 (1): 124—5. PMID 7813066. Архів view=long&pmid=7813066 оригіналу за 20 листопада 2011. Процитовано 25 липня 2019.
- ↑ Kane, Kimberly (16 жовтня 2012). Зак любить Манді, і вони будуть продовжувати робити порно. Архів оригіналу за 26 вересня 2016. Процитовано 28 березня 2022.
{{cite web}}: Cite має пустий невідомий параметр:|7=(довідка) - ↑ Але ви не виглядаєте хворими: життя з хронічним болем (синдром Елера-Данлоса). YouTube. Архів оригіналу за 3 березня 2010. Процитовано 2014-02- 27.
- ↑ "Люди залишаються гнучкими": хворі на рідкісні хвороби відчувають, що їх не помічають медичні служби last = Баррі. TheJournal.ie. Архів оригіналу за 18 березня 2022. Процитовано 25 лютого 2016.
{{cite web}}:|first=з пропущеним|last=(довідка) - ↑ Carrie (26 грудня 2016). Queer Crip Love Fest: Розмовляючи з активістом Latinox Енні Сегаррою про сім'ю. Autostraddle. Архів оригіналу за 23 вересня 2017. Процитовано 22 вересня 2017.
- ↑ Gillibrand, Abigail (1 березня 2019). Jameela Jamil reveals battle with EDS 3 and is proud of ‘overcoming its hurdles’. MetroUK. Associated Newspapers Limited. Архів оригіналу за 2 березня 2019. Процитовано 4 березня 2019.
- ↑ Архівована копія. Архів оригіналу за 1 квітня 2019. Процитовано 29 квітня 2019.
{{cite web}}: Обслуговування CS1: Сторінки з текстом «archived copy» як значення параметру title (посилання) - ↑ Dokuzeylül, Banu; Altun, Elif Demet; Özdoğan, Tamer Halit та ін. (2013). Cutaneous asthenia (Ehlers–Danlos syndrome) in a cat. Turkish Journal of Veterinary and Animal Sciences. 37 (2): 247. doi:10.3906/vet-1203-64.
- ↑ Scott, Danny W (2008). Congenital and hereditary skin diseases. Color Atlas of Farm Animal Dermatology. Wiley Online Library. с. 61. doi:10.1002/9780470344460. ISBN 9780470344460.
- ↑ Halper, Jaroslava; Kim, Byoungjae; Khan, Ahrar; Yoon, Jung; Mueller, PO (2006). Degenerative suspensory ligament desmitis as a systemic disorder characterized by proteoglycan accumulation. BMC Veterinary Research. 2: 12. doi:10.1186/1746-6148-2-12. PMC 1459153. PMID 16611357.
{{cite journal}}: Обслуговування CS1: Сторінки із непозначеним DOI з безкоштовним доступом (посилання) - ↑ Kim, Byoungjae; Yoon, Jung Hae; Zhang, Jian; Eric Mueller, P.O.; Halper, Jaroslava (2010). Glycan profiling of a defect in decorin glycosylation in equine systemic proteoglycan accumulation, a potential model of progeroid form of Ehlers–Danlos syndrome. Archives of Biochemistry and Biophysics. 501 (2): 221—31. doi:10.1016/j.abb.2010.06.017. PMID 20599673.
Сабреддіт (англ.) | |
| Наука | |
| Тематичні сайти | |
| Словники та енциклопедії | |
| Довідкові видання | |
| Нормативний контроль |