
Mucopolissacaridose
| Mucopolissacaridose | |
|---|---|
 | |
| Especialidade | endocrinologia |
| Classificação e recursos externos | |
| CID-10 | E76 |
| CID-9 | 277.5 |
| CID-11 | 1596128696 |
| OMIM | 252700 |
| MedlinePlus | 001246 |
| MeSH | D009083 |
Mucopolissacaridose ou MPS é um subgrupo das doenças de depósito lisossômicos (DDL) as quais pertencem ao ainda maior grupo de doenças genéticas do metabolismo, causadas por deficiência de enzimas. Juntas, afetam cerca de 1 em cada 25.000 nascidos vivos por ano.[1]
Causa
[editar | editar código-fonte]Nas MPS, existe a deficiência ou falta de uma determinada enzima nos lisossomos, o que leva ao acumulo de glicosaminoglicanos (GAG), conhecida antigamente como Mucopolissacarídeos, nome que deu origem a patologia. Os glicosaminoglicanos são moléculas que possuem em sua composição açucares que se ligam a uma proteína central. Essa molécula absorve água em demasia, adquirindo uma consistência viscosa, promovendo assim a lubrificação entre os tecidos, permitindo o deslizamento na movimentação entre eles. Essa diminuição de atrito entre os tecidos permite, por exemplo, o movimento das articulações ósseas. Esse acumulo leva a disfunção na lubrificação dos órgãos causando danos progressivos.[2]
Sinais e sintomas
[editar | editar código-fonte]As manifestações clínicas das MPS são normalmente multissistêmicas (afetam diversos órgãos) e muito variáveis, existindo formas leves, moderadas e graves. Podem afetar o cérebro, olhos, ouvidos, coração, fígado, ossos e articulações.
Genética
[editar | editar código-fonte]É estimado que 1 em cada 25.000 bebês nascidos nos Estados Unidos possuem alguma forma de Mucopolissacaridose. A maioria das MPS são de herança autossômica recessiva, ou seja, apenas indivíduos que herdam o gene defeituoso de ambos os pais são afetados. (A exceção é a MPS tipo II, conhecida também como Síndrome de Hunter, em que a mãe sozinha passa o gene defeituoso para o filho.) Quando tanto o pai, quanto a mãe possuem o gene defeituoso, cada gravidez carrega consigo uma chance de 1 em 4 de que a criança seja afetada. Os pais e os irmão de uma criança afetada podem não ter sinais da doença. Irmãos não afetados e parentes selecionados de uma criança com um dos tipos de Mucopolissacaridose podem portar o gene recessivo e transmiti-lo a seus próprios filhos.
-
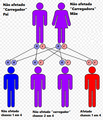 A mucopolissacaridose possui um padrão autossômico recessivo de herança.
A mucopolissacaridose possui um padrão autossômico recessivo de herança.

Tipos
[editar | editar código-fonte]Existem doze tipos de MPS atualmente descritas, que são classificadas de acordo com o tipo de enzima deficiente na célula. A MPS Plus é o tipo mais recentemente descrito de MPS[3] e se distingue por não ser decorrente de uma deficiência enzimática.
| Tipo | Nome da síndrome | Enzima deficiente / Proteína afetada | Produtos acumulados | Sintomas | Localização do gene |
|---|---|---|---|---|---|
| MPS I | Síndrome de Hurler
(anteriormente chamada de MPS V) |
α-L-iduronidase |
|
|
4p 16.3 |
| MPS II | Síndrome de Hunter | Iduronato sulfatase |
|
(sintomas similares a síndrome de Hurler, só que mais brandos) |
Xq28 |
| MPS III | Síndrome de Sanfilippo A | Heparan sulfato sulfatase | sulfato de heparan |
|
17q25.3 |
| Síndrome de Sanfilippo B | N-acetilglucosaminidase | sulfato de heparan | 17q21 | ||
| Síndrome de Sanfilippo C | Acetil-CoA:alfa-glucosaminideo
N-acetil transferase |
sulfato de heparan | 12q14 | ||
| Síndrome de Sanfilippo D | N-acetilglucosamina 6-sulfatase | sulfato de heparan | 8p11 | ||
| MPS IV | Síndrome de Morquio A | Galactose 6-sulfatase |
|
|
16q24.3 |
| Síndrome de Morquio B | Beta-galactosidase 1 |
|
3p22.3 | ||
| MPS VI | Síndrome de Maroteaux-Lamy | N-acetilgalactosamina 4-sulfatase |
|
|
5q11-13 |
| MPS VII | Síndrome de Sly | β-glucuronidase |
|
|
7q21.11 |
| MPS IX | Deficiência de Hialuronidase | Hialuronidase | Hialuronan |
|
3p21 |
| MPS Plus[3] | Síndrome mucopolissacaridose
plus |
Proteína VPS33A | sulfato de heparan |
|
12q24 |
Todas os tipos de MPS, exceto o tipo II são herdados de maneira autossômica recessiva, enquanto a tipo II tem padrão de herança ligado ao cromossomo X.
Casos de Mucopolissacaridose no Brasil
[editar | editar código-fonte]No Brasil existem mais casos de Mucopolissacaridose do que se imagina, contabilizado 600 pessoas. De acordo com a Associação Paulista dos Familiares e Amigos dos Portadores de Mucopolissacaridose, no estado de São Paulo estima-se que existam 150 casos confirmados. De acordo com registros nas associações para portadores de Mucopolissacaridose, Pernambuco é o 2º estado que mais registra casos de mucopolissacaridose.
Associação para Portadores de Mucopolissacaridose no Brasil
[editar | editar código-fonte]No Brasil existem duas principais Associações para portadores de Mucopolissacaridose. Essas associações servem para ajudar legal e psicologicamente os familiares e portadores de mucopolissacaridose. Dentre todas, as principais são a de São Paulo, o Instituto Vidas Raras e em Santa Catarina.
Tratamento
[editar | editar código-fonte]Assim como para muitas doenças genéticas raras, não existe cura para as mucopolissacaridoses, mas existem tratamentos para melhorar a qualidade de vida dos pacientes. Alguns tratamentos são feitos a base de fármacos, numa tentativa de realizar as funções deficientes pelas enzimas que faltam.
Entre os tratamentos mais utilizados e eficazes está a Terapia de Reposição Enzimática (TRE). A TRE consiste em introduzir via venosa o fármaco deficiente nos lisossomos das células. Até o momento há produtos de terapia enzimática aprovados para administração em pacientes com MPS I, MPS II, MPS IV A, MPS VI e MPS VII. Um exemplo é a laronidase, utilizada para a Mucopolissacaridose tipo I.
Além disso, o transplante de células-tronco hematopoiéticas (ou transplante de medula óssea - TMO) é outro tratamento bem estabelecido e eficaz para crianças com a forma grave de MPS I, tendo também sido utilizado com sucesso em alguns casos de MPS II, MPS IVA, MPS VI e MPS VII.[5] O objetivo do transplante é fornecer células de um doador saudável que, após se multiplicarem, produzem quantidades suficientes da enzima deficiente.
Ver também
[editar | editar código-fonte]Referências
- ↑ «Cópia arquivada». Consultado em 22 de julho de 2014. Arquivado do original em 28 de julho de 2014
- ↑ «Cópia arquivada». Consultado em 22 de julho de 2014. Arquivado do original em 18 de agosto de 2016
- ↑ a b c Kondo, Hidehito; Maksimova, Nadezda; Otomo, Takanobu; Kato, Hisakazu; Imai, Atsuko; Asano, Yoshihiro; Kobayashi, Kaori; Nojima, Satoshi; Nakaya, Akihiro (1 de janeiro de 2017). «Mutation in VPS33A affects metabolism of glycosaminoglycans: a new type of mucopolysaccharidosis with severe systemic symptoms». Human Molecular Genetics. 26 (1): 173–183. ISSN 1460-2083. PMID 28013294. doi:10.1093/hmg/ddw377
- ↑ «The Mucopolysaccharidoses | The Online Metabolic and Molecular Bases of Inherited Disease | OMMBID | McGraw-Hill Medical». ommbid.mhmedical.com. Consultado em 25 de julho de 2019
- ↑ Poswar, Fabiano; Baldo, Guilherme; Giugliani, Roberto (2 de dezembro de 2017). «Phase I and II clinical trials for the mucopolysaccharidoses». Expert Opinion on Investigational Drugs (em inglês). 26 (12): 1331–1340. ISSN 1354-3784. doi:10.1080/13543784.2017.1397130
- Neufeld E.F., Muenzer J. In: The Metabolic and Molecular Bases of Inherited Disease. 2001: 3421–3452.
- Meikle P. et al. JAMA. 1999; 281:249-254.