

Abstract
Phages have highly compact genomes with sizes reflecting their capacity to exploit the host resources. Here, we investigate the reasons for tRNAs being the only translation-associated genes frequently found in phages. We were able to unravel the selective processes shaping the tRNA distribution in phages by analyzing their genomes and those of their hosts. We found ample evidence against tRNAs being selected to facilitate phage integration in the prokaryotic chromosomes. Conversely, there is a significant association between tRNA distribution and codon usage. We support this observation by introducing a master equation model, where tRNAs are randomly gained from their hosts and then lost either neutrally or according to a set of different selection mechanisms. Those tRNAs present in phages tend to correspond to codons that are simultaneously highly used by the phage genes, while rare in the host genome. Accordingly, we propose that a selective recruitment of tRNAs compensates for the compositional differences between the phage and the host genomes. To further understand the importance of these results in phage biology, we analyzed the differences between temperate and virulent phages. Virulent phages contain more tRNAs than temperate ones, higher codon usage biases, and more important compositional differences with respect to the host genome. These differences are thus in perfect agreement with the results of our master equation model and further suggest that tRNA acquisition may contribute to higher virulence. Thus, even though phages use most of the cell’s translation machinery, they can complement it with their own genetic information to attain higher fitness. These results suggest that similar selection pressures may act upon other cellular essential genes that are being found in the recently uncovered large viruses.
Parasites face numerous problems when colonizing their hosts. First among these is the optimization of host exploitation, which is a particularly important problem for obligatory lethal pathogens, because they have to get by with the available host resources. Host exploitation is often difficult to study because it is linked with complex traits. This is one of the reasons why the antagonistic associations between bacteria and their phages have recently resurged as interesting models to understand host-pathogen interactions and resulting life-history traits (Turner and Chao 1999; De Paepe and Taddei 2006). Phages are also important shuttles of horizontal gene transfer, and thus, major elements in the dynamics of bacterial evolution (Casjens 2003; Canchaya et al. 2004; Daubin and Ochman 2004). Among the genes carried by phages, toxins are particularly important for bacterial pathogenicity (Waldor and Mekalanos 1996). Thus, phages and bacteria can transiently establish mutualistic interactions to antagonize eukaryotic hosts. Since phages are the most abundant life form on earth (Suttle 2005), the consequences of their ecological interactions are most relevant for both the global ecosystem and human health.
The genomes of phages are typically small, providing them with few tools to divert resources from their environment. Accordingly, they have no proper metabolism and rely on the host cell’s materials for their reproduction (Weinbauer 2004). Phages also rely on the host machinery to reproduce, and while some code for their own RNA and DNA polymerases (Knopf 1998), they require most of the cell’s translation apparatus for protein synthesis. Accordingly, the biases operating in the host sequences toward translation optimization may also operate in the phage genome. It has even been supposed (Krakauer and Jansen 2002) that these biases could be a major force of both phage and bacterial genomes co-evolution.
Selection for optimal codon usage plays a major role in shaping bacterial genomes (Andersson and Kurland 1990), especially in fast-growing bacteria (Rocha 2004; Sharp et al. 2005) and among highly expressed genes (Grantham et al. 1981). In cellular organisms, the optimal codon usage is typically the one fitting best the abundance of tRNAs in the cell under exponential growth conditions (Ikemura 1981). Thus, there is a co-evolution of tRNA abundance and codon usage bias that shapes the abundance of the different codons in gene sequences through long periods of time. This trait is under selection but is counteracted by the action of random mutations that tend to make codon usage bias a reflection of the extant mutational biases (Muto and Osawa 1987). Codon usage bias is thus said to be under selection-mutation-drift balance (Bulmer 1991), and the result is that the bias is more intense (1) in the genes under stronger selection, which are often, but not always (Elf et al. 2003; Bailly-Bechet et al. 2006) the most highly expressed (Gouy and Gautier 1982), and (2) in those moments when there is more selection for the trait, e.g., exponential growth for the most highly expressed genes (Dong et al. 1996). When a phage propagates in a bacterium cell, it is convenient to have a codon-usage bias compatible with the one of the bacteria, as that will facilitate the expensive and laborious mechanism of protein synthesis. Yet, this may be impossible to achieve because phages share with other parasitic DNA a tendency to be A+T rich relative to their bacterial hosts (Rocha and Danchin 2002). The gap between phage and host genome compositions makes it difficult for the phage to perfectly fit the host codon usage.
Nearly 40 years ago, it was found that T4 phages carry some tRNA genes (Weiss et al. 1968). Although deletion of these genes leads to lower burst sizes and rates of protein synthesis (Wilson 1973), the reasons why some phages contain tRNAs have remained enigmatic. Early work, also on T4, suggested that its tRNA gene content corresponded to the codon usage of some lowly expressed genes in the phage, for which the corresponding host tRNAs were too rare (Cowe and Sharp 1991; Kunisawa 1992). These studies also found that, while highly expressed genes in T4 tend to have a codon usage bias close to the host, Escherichia coli, lowly expressed and late genes use codons for which some of the eight T4 tRNAs could be useful. Yet, at that time, the lack of data did not allow one to understand whether the observation was a peculiarity of T4 and E. coli or a general feature. The finding that phages closely related to T4 showed extensive polymorphisms in the number and type of tRNA genes contributed to the near abandonment of the work on this hypothesis (Miller et al. 2003). Meanwhile, the availability of hundreds of bacterial genomes highlighted the role of phages in bacterial evolution as vectors of horizontal gene transfer. About half of the sequenced genomes contain prophage sequences (Canchaya et al. 2004) and these may constitute up to 16% of the genome (Ohnishi et al. 1999). Importantly, prophage integration occurs at a tRNA gene for phages carrying lambda and P4-like integrases (Campbell 1992). These phages do not carry tRNAs, but only a small part of a tRNA that compensates for the disruption in the host tRNA. In this context it has been proposed that tRNA presence in phages could be a by-product of imprecise excision of prophages. This would not strictly require a positive effect of tRNAs on phage fitness, although one might suppose that their presence could be selected to compensate for insertions inactivating the host’s tRNA (Canchaya et al. 2004). A problem with this hypothesis is that it fails to explain the presence of tRNAs in nontemperate phages. A possible explanation would be the capture of bacterial DNA by the virulent phage during host chromosome degradation, before encapsulation of all of the phage genetic material, as this process liberates large quantities of DNA (Weinbauer 2004). Virulent phages could also acquire tRNAs by recombination with temperate phages. As genomic data showed that tRNAs provide integration points for phages, plasmids, and pathogenicity islands, other putative roles for phage-encoded tRNAs have been neglected. Yet, this issue gains a special relevance in light of the important role of phages in bacterial evolution. It may also help understand the unexpected recent discovery that large eukaryotic viruses contain other elements of the translation machinery, such as elongation factors and tRNA synthetases (Raoult et al. 2004).
We have thus decided to investigate the relationship between tRNA copy number and codon frequency in bacteriophage genomes relative to their hosts. This is now possible because hundreds of phage and bacterial genomes have been sequenced. The study of codon usage in bacteriophage genomes presents additional constraints relative to the equivalent study in bacteria. Firstly, the process of selection of tRNAs must take into account that tRNAs have probably been taken from the host genome. Secondly, the codon bias of phages is modified by a general compositional bias toward higher A+T content than the host genome, which could blur the simple picture that arises in bacteria for the relationship between tRNA content and codon usage. Finally, the low number of tRNAs present in phage genomes implies the usage of careful statistics and sophisticated models. We have identified tRNAs in phage genomes and in their hosts and investigated the correlation between the tRNAs of the phage and its codon bias. Next, we developed a master equation model to simulate the acquisition and loss of tRNAs by phages, and, using likelihood comparisons, we found the most relevant selective processes that could drive it. We finally placed these results in the framework of phage ecology and evolution.
Results
Associations between tRNAs and codon usage in phages and their hosts
We collected from GenBank the complete genomes of phages and their hosts. We then identified tRNAs in both groups of genomes, removing some elements that could complicate our analysis, such as pseudogenes (see Methods). We eliminated from further analyses all phages that were not annotated, which lacked tRNAs, and those without a completely sequenced bacterial host. The final data set contains 15 host bacterial genomes and 37 genomes of phages. The genomes of these phages contain a total of 169 tRNAs, thus showing an average of approximately four tRNAs per phage. However, such an average value is somewhat misleading, since the number of tRNAs inside each phage is very variable (Fig. 1). Most phages contain only one or two tRNAs, while a few contain more than 20 such sequences, which is nearly as many tRNAs as one can find in bacteria with minimal genomes (Rocha 2004). All tRNA-containing phages are dsDNA phages. The main difference between the phages with tRNAs and those without any tRNAs stands in genome length: phages containing tRNAs are significantly longer than those without (average lengths are, respectively, 74 and 32 kb, P = 10−6). We then separated phages into virulent and temperate, according to the published information on their ecology. In some cases both annotation files and published literature lacked information allowing such a classification and those phages were thus gathered in a third group. We could classify 21 phages as temperate and 12 as virulent. The abundance of tRNAs in their genomes is very different (Fig. 1), with no temperate phage containing more than four tRNAs. We shall get back to this issue in a subsequent section as it is relevant to understanding the role of tRNAs in phages.
Figure 1.

Distribution of the number of tRNAs inside phage genomes. Empty bars stand for temperate phages, filled bars for virulent phages, and patterned bars for phage of unknown type. Note the heterogeneity of the counts and the tendency for virulent phages to have more tRNAs than temperate ones. Names are indicated for phages with 19 or more tRNAs in the form “(host species) Phage.”
We started our analyses by testing the hypothesis that the tRNA gene content for each anticodon is positively correlated with the complementary codon frequency in the genome of phages. This would be a situation similar to the one of bacteria, where tRNA gene content is highly correlated both with tRNA cellular content (Ikemura 1985; Dong et al. 1996) and codon frequency (Ikemura 1981). The test performed in each phage independently shows weak statistical power due to the few tRNAs they contain. Hence, we took all 37 phage genomes into account and computed the probability distribution of their codon frequencies, i.e., the probability p(f) that any codon inside a phage will be used at a given frequency f. One can see that this distribution does not differ between the phages and their hosts (Fig. 2). This means that, on average, few codons are highly used, while most are rare, and that the trends are similar in phages and their hosts. Then, we computed the same probability distribution restricted to the codons for which at least one cognate tRNA is found in the bacteriophage genome. The curve is rougher because of the lower number of points, but it is clearly different from the previous one (Kolmogorov-Smirnov test, P < 10−3). The peak around f = 0.03 is due to the presence of numerous tRNAMet inside phage genomes, in which methionine appears to be often used around this frequency. In the inset of Figure 2, we plot the cumulated probability distribution of the codon frequency, i.e., the probability of finding codons with frequencies superior to the value given on the X-axis, for phage codons with and without the complementary tRNA. The lag between the two curves shows that there is a high proportion of codons having a matching tRNA among the high-frequency codons. It is thus clear that there is a positive association between the frequency of a codon and the presence of the cognate tRNA in the phage genome.
Figure 2.

Distribution of the frequencies of codon usage in phage genomes. The solid line is the distribution of codon frequencies; the dotted line, the distribution of codon frequencies, restricted to codons matching a tRNA on the considered phage genome. Note the peak around f = 0.03. The dash–dot line is the frequency distribution for all codons of all hosts. In the inset, the cumulated probability distribution (probability that a random tRNA will have a frequency superior or equal to the one given in abscissa) of the tail of the frequency distributions of the phages for all codons (solid line), and only the ones matching a tRNA (dotted line). Note the difference, indicating an excess of tRNAs matching high-frequency codons in phages.
We then computed the correlations of codon frequencies between each phage and its host. As previously noted for some of these phages (Sharp et al. 1985; Kunisawa et al. 1998; Kropinski and Sibbald 1999; Sau et al. 2005), we find highly correlated codon frequencies: the average of the Pearson’s coefficient R on all 37 couples phage-host is 0.78 ± 0.04 (standard error), and 36 of the 37 associated P-values are inferior to 0.05. This value has to be compared with the value found by computing the average correlation coefficient, 0.38 ± 0.02, between a phage and a random host (computed between the phage and a random bacterial genome from the 356 we found available in GenBank). We conclude that the frequencies of codon usage are very correlated between a phage and its host. A similar pattern of correlations is observed among phages containing no tRNAs.
If phages and their hosts had exactly the same codon usage, then a trivial explanation of the results of the previous paragraph would be that phages pick tRNAs randomly from their hosts and that the correlation between phage codon usage and tRNA gene content simply reflects the association between tRNA abundance and codon usage in the host. No hypothesis about selection on tRNAs in phages would then be required. Yet, a coefficient of correlation of 0.78 only allows explaining about half of the variance, leaving ample room for an autonomous selection strategy of tRNA acquisition in phages. We shall show that the no-selection hypothesis does not fit the data as adequately as some models featuring selection.
tRNAs may be randomly recruited from the hosts, but they are selectively kept
One can explain the previous results in at least two different ways. A purely neutral hypothesis is that tRNAs are drawn at random from the host genome. A selective refinement of this hypothesis is that tRNAs are drawn at random and kept because they help phage integration. In this case, tRNAs should be kept in the phage at the same frequency as in the hosts, and their distribution would differ from a random uptake with no selection only by a greater magnitude of the rate of tRNA acquisition.
A second alternative is that, after tRNA recruitment, there is selection for keeping some tRNAs but not others. One would expect this to be correlated with the phage codon usage or the difference in codon usage between the phage and the host. In the first case, the tRNAs would render the phage less dependent on the host to translate its own proteins. The second case implies an evolutionary strategy of compensation of codon usage differences, where the phage keeps those tRNAs that are rare in the host and whose cognate codons are frequent in the phage genome.
We first assessed whether it is reasonable to assume that tRNAs are randomly picked from bacterial hosts. When analyzing the anticodons of tRNA genes we only observed eight tRNAs, of 177, that were present in the phage and not in its host. In three cases the Cove score of these tRNAs, as given by tRNAscan_SE, is <30, which is at the borderline of significance and suggests that they are false positives. For the five remaining tRNAs, we checked whether some putative other host of the same phage had the corresponding tRNAs (i.e., a tRNA with the same anticodon). As discussed in the Methods, in these comparisons we only used one randomly chosen host for the phage, even when we knew several (e.g., in multiple sequenced strains of a species). In all of the five cases we did find another host genome containing the phage tRNAs. Thus, all reliable tRNAs we observe are at least present in a given known host, in accordance with our recruitment hypothesis. The phylogenetic signal is erased very quickly. Yet, we aligned the tRNA sequences of the phages with those of all sequenced genomes of the host genus, and computed the average similarity between these sequences. The average over all 169 sequences is 70.74%. Unfortunately, it is impossible to make phylogenetic analyses of the tRNA genes found in phages to check the acquisition from the host, because tRNA genes are very small and once acquired by phages they mutate 100- to 1000-fold faster than in the hosts per generation (Drake 1991), which is further enhanced by the very high growth rates of phages.
We computed for each phage the sum of its codon frequencies weighted by the number of exact cognate tRNAs it contains, and compared it with the value it would take if the same number of tRNAs were drawn at random from the host genome. We observed no significant association between the phage tRNA gene content and its codon usage bias (Kolmogorov test, P = 0.68).
We then computed the sum of the differences in codon usage between phage and host, weighted in the same way. This showed a highly significant association between tRNA presence and codon usage difference: the probability that the observed difference in codon usage arises via random tRNA uptake from the hosts is 4 × 10−3. We conclude that the tRNA gene content in a bacteriophage is not due to a simple random drawing from the host tRNA distribution.
One usually assumes that tRNAs are integrated in the phage genomes either at the time of chromosome degradation or when there is imprecise excision of prophages. If tRNAs are recruited from the host genome without further selection, then one would expect temperate phages to contain more tRNAs than virulent phages (if we suppose their rates of acquisition of free DNA sequences to be equal) because they might use both mechanisms. Yet, virulent phages have more, not less, tRNA genes than temperate phages. Since tRNA genes are not a random sample of the host tRNA genes, we tried to identify the underlying selective process.
Modeling the acquisition and selective loss of tRNAs inside phage genomes
We used a master equation approach to model the putative selective processes involved in the modulation of the phages tRNA gene content (see Methods). More precisely, we modeled the evolution of the probability  αβ,x(n) of phage α (whose host is β) to have n tRNAs of anticodon x. We initially modeled only the random processes of acquisition and loss, i.e., our null model involves no selection upon the tRNA gene content. We defined the rate of acquisition, r, by normalizing the rate of loss to 1 (see Methods). We estimated the value of r by maximum likelihood and found r = 0.063. This low value is in accordance with the fact that most phages have no or few tRNAs, as it coincides with the relative frequency of tRNAs in phages relative to their hosts.
αβ,x(n) of phage α (whose host is β) to have n tRNAs of anticodon x. We initially modeled only the random processes of acquisition and loss, i.e., our null model involves no selection upon the tRNA gene content. We defined the rate of acquisition, r, by normalizing the rate of loss to 1 (see Methods). We estimated the value of r by maximum likelihood and found r = 0.063. This low value is in accordance with the fact that most phages have no or few tRNAs, as it coincides with the relative frequency of tRNAs in phages relative to their hosts.
Absence of selection of tRNAs for phage or host codon usage
We then proceeded to introduce in the model different processes to identify the one that brought significant information. We specifically explored the relevance of three different selection processes to explain tRNA composition in phages. We started by considering the hypothesis that tRNAs are selected to match the most abundant codons in the phage genome. For this, we specified a rate of tRNA loss decreasing in fα,x (the frequency of codon usage of phage α for codon x) and controlled by a selection coefficient s. We solved the new master equation, and found r and s by the maximum of the log-likelihood  . The significance of a non-zero value of s can be computed by comparing the log-likelihood r for the null model with s = 0, i.e., no selection, and the log-likelihood r,s for the model with selection. The likelihood ratio method indicated that our estimation of s is not significantly different from 0 (P = 0.15), suggesting little, if any, tRNA selection based on the phage codon usage bias.
. The significance of a non-zero value of s can be computed by comparing the log-likelihood r for the null model with s = 0, i.e., no selection, and the log-likelihood r,s for the model with selection. The likelihood ratio method indicated that our estimation of s is not significantly different from 0 (P = 0.15), suggesting little, if any, tRNA selection based on the phage codon usage bias.
We then tested whether phage tRNAs could be selected to match those codons that are rarer in the host. The estimated s value for this process was significant (P = 0.018), but only before applying the Bonferroni correction for our multiple statistical tests (P = 0.072). This selection process was therefore also discarded.
Our initial exploration of tRNA composition in phages showed very few cases of duplicated tRNA genes, since we only found six pairs and one triplet of similar tRNA species in a phage. Nevertheless, we tested explicitly the hypothesis that tRNA gene amplifications may contribute to explaining tRNA distribution in phages by simulating a process where tRNAs can be duplicated at a rate c. We considered too unlikely the hypothesis that a duplicated tRNA could mutate to become another species, because this could lead to a tRNA present on the phage and not in its host, which is not observed. The duplication rate, c was then estimated by maximum likelihood, but we inferred c = 0 as the most probable value. Hence, the most likely scenario for the multiple acquisition of similar tRNA species is the one of independent rounds of tRNA acquisition, not of gene duplication.
Selection for differences in codon usage between phage and host
We finally tested the hypothesis that selection is based on the difference between the frequency of codons in the phage and its host. Our rationale was that selection could favor phages with tRNAs corresponding to codons that are abundant in the phage but rare in the host. These codons are expected to be poorly translated by the host machinery and lead to slow phage growth if not compensated by the phage’s own tRNAs. Solving the new model by maximum likelihood, we found s to be significantly different from zero even with the correction for multiple tests (P < 2 × 10−7). This strongly suggests that the selection process acting on tRNAs is based on the difference of codon usage between host and phage.
To further validate this conclusion we made three additional tests. Firstly, we solved the master equation model with a randomized data set, generated by taking the observed values of the tRNA counts and associating them to random codon frequencies of the phage and hosts taken among those observed. This randomization deleted the internal correlation between host and phage codon frequencies, and phage tRNA counts. As expected, the model gave nonsignificant values of the selection coefficient s when applied on this data set.
Secondly, since most phages contain very few tRNAs, we used a binary model where αβ,x(n) can only have the value 0, for no tRNA of a given type in the phage, and 1, for at least one such tRNA in the phage (see Methods). This binary model leads to some loss of information but is expected to be more robust. We found a similar P-value for the rejection of the hypothesis that s is equal to 0 (P < 5 × 10−9). Thus, results seem robust and give strong support to the hypothesis that tRNAs are selected in phages to compensate for differences in codon usage between the phage and the host.
Finally, we tested whether our results are robust to changes in the arbitrary selection of one among several known hosts. We made the same analysis by assuming each phage to infect all strains of a same species. This was done by creating an average genome representing the species genome (see Methods). Both the statistical analysis and the master equation modeling gave qualitatively and quantitatively the same results, highlighting their robustness.
Higher abundance of tRNAs in virulent phages
Virulent and temperate phages have very different ecologies. Thus, we investigated whether both groups used tRNAs to compensate for differences in codon usage with respect to the host. For this, we fitted the master equation model (without the nonsignificant gene duplication term) to the two sets of phages separately.
Splitting the phages leads to groups with low effectives. Yet, we still found a significant effect of selection for tRNA genes caused by the difference of codon usage between host and phage Δfαβ,x, both for virulent and temperate phages (resp. P < 5 × 10−7 and P < 5 × 10−4). Thus, both types of phages contain tRNAs corresponding to their mid- to high-frequency codons (Fig. 3), which are also those showing the largest difference to the host codon usage.
Figure 3.

Distribution of the frequencies of codon usage in virulent phage genomes (top) and temperate phage genomes (bottom). Light-gray filled bars, the distribution of codon frequencies, for all codons; black empty histogram bars, the distribution of codon frequencies, restricted to codons matching a tRNA on the considered phage genome. Note the difference between the distributions in both the virulent and the temperate case.
Even if selection is present in both types of phages, the significance is higher for virulent phages, in spite of their smaller sample size. This is corroborated by three other observations. Firstly, in our sample, virulent phages have an average of 7.9 tRNAs, whereas temperate phages only have 2 tRNAs (Fig. 1, significant difference, P < 3 × 10−3). Secondly, the codon usage of the hosts correlates much better with the one of temperate phages (0.83 ± 0.03) than with virulent phages (0.61 ± 0.11). Thirdly, virulent phages tend to have stronger codon usage bias. To quantify this assumption, we used a measure of deviation of codon usage from a uniform distribution that accounts for the nucleotide composition of the genome,  (Novembre 2002). We computed its value for each phage and found that virulent phages are significantly more biased than temperate (Wilcoxon test, P < 5 × 10−3).
(Novembre 2002). We computed its value for each phage and found that virulent phages are significantly more biased than temperate (Wilcoxon test, P < 5 × 10−3).
Temperate and virulent phages are distinct in one major aspect. Temperate phages replicate both through lytic cycles and in the lysogenic state, while in the latter, temperate phages share the same mutational biases as the host. As a result, they tend to have a genome composition much closer to the one of the host than virulent phages, which share with other parasitic DNA a bias toward A+T richness relative to the host (Rocha and Danchin 2002). This could lead temperate phages to show a codon usage bias closer to the one of the hosts, as observed. Since there are more differences in codon usage between the host and virulent phages, one would expect the latter to contain more tRNAs, in order to compensate for this difference.
Discussion
Bacteriophages have highly compact genomes that tend to lack any translation-associated genes, with the notable exception of tRNAs. Our investigation on the reasons motivating this exception have shown that many genomes lack or have few tRNAs, whereas some genomes contain nearly as many tRNAs as some bacteria. There is a positive association between the size of the phage genome and the number of tRNA genes it contains. This suggests that tRNA genes are part of the phages accessory genome probably arising from multiple recruitment events and only being kept when selection for their presence is strong enough. We only found tRNAs in the genomes of dsDNA phages. The other phages may miss tRNAs either because they are much more compact, thus excluding nonessential information and/or because the folding of tRNAs may pose problems in the organization of the chromosomes of RNA or ssDNA phages. The presence of tRNAs among dsDNA phages is coherent with the following evolutionary scenario. Firstly, tRNAs are recruited from the host chromosomes or from recombination with other phages coinfecting a bacterial cell. Secondly, these tRNAs are subject to frequent deletion following the deletional bias that is thought to predominate in the genomes of bacteria and phages (Lawrence et al. 2001; Mira et al. 2001). Yet, some tRNAs can provide for such an advantage as to counteract the effect of the deletion bias. As long as the advantage of carrying the tRNA overcomes the negative effect of increasing genome size and the deletion bias, the tRNA will be kept in the genome.
Our data indicate that tRNAs that are kept in phage genomes are those corresponding to codons abundant in the phage and rare in the host. This allows the phage to gain a clear-cut advantage over its competitors by translating its proteins more efficiently, reducing its latency time, and increasing the reproduction rate. This may be balanced by the time necessary to replicate the tRNA sequence on the phage genome, but the latter effect must be weak, since tRNA genes are very small.
For a phage to carry and express a tRNA that is already abundant in the host would give little benefit, since it would have a small relative effect on the phage environment during infection. Instead, expressing a tRNA that is rare in the host may provide a decisive benefit to the phage if it corresponds to a highly frequent codon in its own genes. So, the optimal configuration for a phage would be to carry tRNAs matching the codons it uses much more than its host, which is indeed what we observe.
It might be argued at this stage that the best strategy for the phage would be to perfectly mimic the host codon and tRNA usage. Although we did observe significant correlations between the codon usage bias of the phage and its host, this strategy may not be perfectly attainable for two reasons. Firstly, tRNA concentration and codon usage bias in bacteria vary with the physiological state of growth, and in fast-growing bacteria they are mostly determined by the physiological requirements of the exponential phase (Kurland 1991; Dong et al. 1996). These are not the conditions prevailing during the lytic cycle. Secondly, the genomes of phages tend to be AT richer than the genomes of their hosts, which necessarily affects codon usage. The reasons for this bias may be mutational, error-prone polymerases, or inefficient repair, or selective adaptation to the AT richness of the bacterial cytoplasm. In any case, they are more important for virulent than for temperate phages (Rocha and Danchin 2002). As a result, phage codon usage cannot perfectly fit the host translation machinery and the recruitment of the necessary tRNAs becomes adaptive. Such an effect will be more important if the compositional gap is important, if the latency times are shorter, if the phage codon usage bias is higher, and if the phage depends exclusively on horizontal transmission to reproduce. These conditions are met for virulent phages, which, accordingly, contain more tRNAs than temperate ones.
We observed higher codon usage bias in virulent phages. Why should that be? We speculate that it is because virulent phages replicate faster and need to translate very efficiently their mRNAs. Although we could not find data on latency times (the average time it takes a phage to lyse the host after infection) for most our phages, we did find a recent work describing these values for some E. coli phages (De Paepe and Taddei 2006). When comparing these latency times for dsDNA phages, we found a statistically significant difference, with lower values for virulent phages (28 vs. 54 min on average, P < 0.02, Wilcoxon test). Thus, increased codon usage bias in virulent phages might result from selection for lower latency times. Virulent phages would then tend to select more strongly for the presence of tRNA genes, both because they are more at odds with the host codon usage and because they are under stronger selection for codon usage bias.
Other models have been put forward to explain the presence of tRNAs among phage genomes, e.g., models where the presence of tRNAs allows the phage to be resistant to anticodon nucleases in the host (Kaufmann 2000; Blanga-Kanfi et al. 2006), the use of alternative genetic codes (see, for example, Bacher et al. 2003), or a better integration of lysogenic phages inside the host chromosome (Canchaya et al. 2004). The first two hypotheses are based on very few observations, and it is still unclear whether they are indeed strategies to evade host response and whether they are frequently found in nature. The last model is contradicted by our observation that lytic phages contain more tRNAs than temperate ones and that the populations of tRNA genes in phages are not random samples of the host repertoire. Moreover, most known temperate phages inserting in a tRNA gene of the host genome (e.g., E. coli phage P22, P4, or Lambda) have no tRNA genes. This shows that these genes are not necessary for phage integration in the bacterial chromosome. In contrast, our model is grounded on the well-known advantage of carrying tRNA genes for translation optimization of the cognate codons and was confirmed by several different tests and controls.
Phages are a major vehicle of lateral gene transfer in bacteria. However, tRNA genes are essential housekeeping and information-related genes, which are expected to be the least prone to horizontal gene transfer (Jain et al. 1999). Their occurrence in phages may lead to the lateral transfer of tRNAs from one cellular genome to another, but our data indicate that their presence is much more likely to be caused by the advantage that they confer to phages in hosts depleted in these tRNAs. Recently, a wealth of viruses and phages with large genomes has been discovered, highlighting the potential diversity in terms of their genome structure and different functionalities of viruses (Ghedin and Claverie 2005). Here, we showed that these may include the recuperation of essential cellular genes from the host to optimize the expression of their own genes in view of infecting those same hosts. Thus, the very large viruses may contain a significant number of translation-associated genes for selective purposes. Surely, the growing number of couples of host/virus complete genomes will reveal the extent and other variants of this evolutionary strategy.
Methods
Data
The genomes of phages and hosts were downloaded from GenBank. The assignment of a phage to a host and to a class of virulence was done using data collected from the literature. Sometimes this classification was impossible because the genes in the GenBank file were not functionally annotated and no information was available in the literature. These phages (four of 37) were used in the generic analysis but were discarded in the comparison between virulent and temperate phages.
The tRNAs sequences in both the phage and the host genomes were detected using tRNAscan_SE (Lowe and Eddy 1997) with default parameters for prokaryotic genomes. We started with a data set of 193 phage genomes, of which 48 contain a total of 214 tRNAs. We excluded from further analysis those tRNAs that could mislead the results of our statistical analysis, i.e., pseudogenes, tRNAs for SeC, undefined tRNAs, and the rare tRNAs absent from the chosen host. We also removed all phages for which no complete sequence of a host was publicly available and those for which there was no annotation. Thus, we used a final data set containing 37 phages corresponding to 15 hosts and including a total of 169 tRNAs. To build the data set of host genomes, we randomly selected for each phage one of its bacterial hosts, when several were fully sequenced, e.g., for E. coli. An alternative analysis using mixtures of several host genomes showed similar results. To test the robustness of the model, statistical tests and the master equation model analysis were also performed on average host genomes. For each host, we randomly chose one genome in each genus of the same family and built an average genome by averaging between all of these genomes the frequencies of codon usage and the tRNA gene content. Tests were then performed considering these genomes as the host representing the putative or unknown wide host range of all phages. We detected tRNAs in the host genomes in the same way. The tRNAs located in the host genomes inside a prophagic region were removed from the pool of data to be analyzed using published information on prophage locations (Canchaya et al. 2004). This avoids including a circularity in the analysis, i.e., comparing tRNAs of phages with those of their prophages. The table with the names and accession numbers of phage genomes and their classification into temperate and virulent are published as Supplemental material.
tRNA alignment
Phage tRNAs were aligned against those of all hosts of the same genus as the chosen host. The alignments were done using the “needle” program (Rice et al. 2000) with a constant gap penalty of 10, which allowed a better alignment of the sequences. Similarities were measured using the same software.
Statistical tests
Comparison of the virulent and temperate phages total tRNA gene content
We used a Monte-Carlo method to test for a statistically significant difference in the number of tRNAs in virulent and temperate phages containing tRNAs. To this aim, we estimated the probability of finding as many tRNAs or more in a group, as observed in the real case. We considered the real distribution of tRNAs counts inside the phage genomes. Here, α denotes a phage, Nα is the number of tRNAs within phage α, and Nα,x is the number of tRNAs having anticodon x in phage α. We drew at random in the {Nα} set 12 values, corresponding to our 12 virulent phages, and sum them. We then estimated the probability distribution of this sum, and computed the probability of having a sum superior or equal to 95, the observed number of tRNAs in the virulent phages.
Statistical test of the random uptake hypothesis
We designed two indicator variables, Aα and Bαβ, that allow testing, respectively, (1) if tRNAs tend to correspond to over-represented codons or (2) if tRNAs correspond to codons used more in the phage than in its host. As a first order approximation, we only considered the correspondence between tRNAs of an anticodon and frequencies of the perfectly matching codons, as these are usually regarded as the optimal codons. We then computed Aα, the average frequency of codon usage restricted to codons for which a matching tRNA is present in the genome, and Bαβ, the average difference in codon usage between the phage and its host, for the same codons. fα,x is the frequency of codon x in phage α, computed on all its genes and relative to all other codons. Δfαβ,x is the difference of the frequencies of codon usage, for codon x, between the phage α and its host β. x is the perfectly matching anticodon for codon x, using Watson-Crick pairing rules. We computed the indicators as:
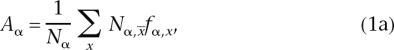 |
 |
To assess the statistical significance of these indicators, we drew at random from the host as many tRNAs as contained in the phage, Nα. By repeating this procedure 100,000 times we obtained the expected distribution of tRNAs in the phage under a model where tRNAs are randomly sampled from the host genome. This allows obtaining the probability  and
and  to have Aα or Bαβ randomly greater or equal to the observed value of the indicators, in each phage. The significant departure of both sets of 37 probabilities
to have Aα or Bαβ randomly greater or equal to the observed value of the indicators, in each phage. The significant departure of both sets of 37 probabilities  and
and  from a uniform distribution was assessed by a Kolmogorov test.
from a uniform distribution was assessed by a Kolmogorov test.
Master equation model
The model
Suppose that the tRNAs of the phage α are taken at random among the tRNAs of its host β, with a rate r supposed to be unique for all phages and anticodons. All tRNAs for different anticodons x are considered independent. Without any lack of generality, we set the rate of loss of the tRNAs to 1 (changing this value would result in the same equation with a rescaling of time t). We denote by Hβ,x the number of tRNAs of host β for anticodon x. The probability αβ,x(n) of phage α having n tRNAs of anticodon x, is governed by the master equation:
 |
where the dependence of αβ,x on time does not appear for easier reading. This initial equation is the mathematical formulation of the hypothesis that all tRNAs present in the genomes of phages are drawn at random from host genomes with a constant rate r, and lost at a rate normalized to unity.
We then added a selection parameter, s, to model how selection changes the probabilities of tRNAs being fixed in the populations of phages. This is achieved by allowing for three different processes that selection acts upon (1) the frequency fα,x of codon x in the phage α; (2) the difference of codon frequencies between the host and the phage, Δfαβ,x = fα,x − fβ,x; (3) the opposite of the frequency fβ,x of codon x in the host genome. The quantity under selection is denoted hereafter by the symbol F (indicating fα,x, Δfαβ,x or −fβ,x for the three cases, 1, 2, and 3, respectively). All of these models are described by the equation:
 |
A positive value of s stands for a selective process tending to keep the tRNAs having a high value of the selected trait F. The exponential form of this selection rate is chosen for simplicity, and since the values of s are small, it is equivalent to using a selection linear in s.
We also considered the hypothesis that a phage tRNA can multiply in the genome with rate c. The master equation corresponding to this case reads:
 |
The stationary solution to Equation 4 can be derived analytically. Here, we show the general solution, with c = 0 or s = 0 being special cases. To find the solution to Equation 4, we first recast the master Equation 4 in terms of the generating function ϕ(λ,t) = ∑n eλnαβ,x(n,t). This gives a new differential equation, which turns out to be of the hypergeometric type and can thus be solved analytically. By applying the opposite transformation from the generating function to αβ,x, we finally obtain the following expression:
 |
One can easily check by direct substitution of Equation 5 into Equation 4 that the right-hand term of Equation 4 indeed vanishes. In the limit c → 0, the previous expression reduces to the expected Poisson law for the stationary probability:
with φ = rHβ,xesF. This solution has the trivial limit φ → rHβ,x as s → 0. In this case, r is the only parameter, and we simply model random acquisition and loss of tRNAs.
Parameter fit
We use a maximum likelihood method to find the most probable values of the parameters r, s, and c. Note that each of the three parameters is supposed to be identical for each phage and anticodon x. Firstly, the log–likelihood of the set of observed counts is computed:
where the dependence on r, s, and c on the left-hand side is brought by the expression (Equation 5). We verified that the log-likelihood landscape is relatively smooth, allowing us to maximize it by simply computing its value at every point of a three-dimensional grid of constant step for each parameter and verifying the maximum thus found by using a steepest gradient method. For very low values of c, which can be computationally tricky, we analytically compute (ln()/∂c)|c=0 and find it always negative in a close neighborhood of the parameters rmax and smax, which maximize ln[(r,s,0)]. This analysis, combined with the absence of solutions found by the other methods for c > 10−10, confirm that the most probable value of c is 0. Then, the two parameters r and s correspond to the zeros of the derivatives of ln[(r,s,0)] relative to them. Computing the derivative of Equation 7 and equating it to zero, gives the relation:
 |
The most probable value of r is the one satisfying Equation 8 and maximizing the log-likelihood (Equation 7). This result also directly gives the value of r when s is taken equal to 0, as in the first model.
The significance of including an additional parameter in the model is computed by the standard likelihood ratio method (Saporta 1990).
Binary model
In the majority of cases, there is only one tRNA for a given anticodon, per phage genome. To confirm the results of the previous model, we designed a simpler two-state model accounting only for presence (+) or absence (–) of tRNAs for a given anticodon. Using the same hypothesis of random uptake and selection as before (c is set to zero in this model), we have:
 |
 |
Equations 9, a and b, are derived and analyzed as previously. The solution of this system is:
 |
 |
In this case, the maximum of the log-likelihood was computed on a grid of precision 10−2 for s, sufficient to demonstrate that s is significantly different from 0.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are online at http://www.genome.org/cgi/doi/10.1101/gr.6649807
References
- Andersson S., Kurland C.G., Kurland C.G. Codon preferences in free-living microorganisms. Microbiol. Mol. Biol. Rev. 1990;54:198–210. doi: 10.1128/mr.54.2.198-210.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher J., Bull J., Ellington A., Bull J., Ellington A., Ellington A. Evolution of phage with chemically ambiguous proteomes. BMC Evol. Biol. 2003;3:24. doi: 10.1186/1471-2148-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly-Bechet M., Danchin A., Iqbal M., Marsili M., Vergassola M., Danchin A., Iqbal M., Marsili M., Vergassola M., Iqbal M., Marsili M., Vergassola M., Marsili M., Vergassola M., Vergassola M. Codon usage domains over bacterial chromosomes. PLoS Comp. Biol. 2006;2:e37. doi: 10.1371/journal.pcbi.0020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanga-Kanfi S., Amitsur M., Azem A., Kaufmann G., Amitsur M., Azem A., Kaufmann G., Azem A., Kaufmann G., Kaufmann G. PrrC- anticodon nuclease: Functional organization of a prototypical bacterial restriction rnase. Nucleic Acids Res. 2006;34:3209–3219. doi: 10.1093/nar/gkl415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell A.M. Chromosomal insertion sites for phages and plasmids. J. Bacteriol. 1992;174:7495–7499. doi: 10.1128/jb.174.23.7495-7499.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchaya C., Fournous G., Brussow H., Fournous G., Brussow H., Brussow H. The impact of prophages on bacterial chromosomes. Mol. Microbiol. 2004;53:9–18. doi: 10.1111/j.1365-2958.2004.04113.x. [DOI] [PubMed] [Google Scholar]
- Casjens S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003;49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- Cowe E., Sharp P.M., Sharp P.M. Molecular evolution of bacteriophages: Discrete patterns of codon usage in T4 genes are related to the time of gene expression. J. Mol. Evol. 1991;33:13–22. [Google Scholar]
- Daubin V., Ochman H., Ochman H. Start-up entities in the origin of new genes. Curr. Opin. Genet. Dev. 2004;14:616–619. doi: 10.1016/j.gde.2004.09.004. [DOI] [PubMed] [Google Scholar]
- De Paepe M., Taddei F., Taddei F. Viruses’ life history: Towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 2006;4:e193. doi: 10.1371/journal.pbio.0040193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H., Nilsson L., Kurland C.G., Nilsson L., Kurland C.G., Kurland C.G. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- Drake J. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. 1991;88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf J., Nilsson D., Tenson T., Ehrenberg M., Nilsson D., Tenson T., Ehrenberg M., Tenson T., Ehrenberg M., Ehrenberg M. Selective charging of tRNA isoacceptors explains patterns of codon usage. Science. 2003;300:1718–1722. doi: 10.1126/science.1083811. [DOI] [PubMed] [Google Scholar]
- Ghedin E., Claverie J.M., Claverie J.M. Mimivirus relatives in the Sargasso sea. Virol. J. 2005;2:62–67. doi: 10.1186/1743-422X-2-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M., Gautier C., Gautier C. Codon usage in bacteria: Correlation with gene expressivity. Nucleic Acids Res. 1982;10:7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R., Gautier C., Gouy M., Jacobzone M., Mercier R., Gautier C., Gouy M., Jacobzone M., Mercier R., Gouy M., Jacobzone M., Mercier R., Jacobzone M., Mercier R., Mercier R. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res. 1981;9:r43–r74. doi: 10.1093/nar/9.1.213-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. Correlation between the abundance of Escherichia coli tRNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 1981;146:1–21. doi: 10.1016/0022-2836(81)90363-6. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Jain R., Rivera M.C., Lake J.A., Rivera M.C., Lake J.A., Lake J.A. Horizontal gene transfer among genomes: The complexity hypothesis. Proc. Natl. Acad. Sci. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann G. Anticodon nucleases. Trends Biochem. Sci. 2000;25:70–74. doi: 10.1016/s0968-0004(99)01525-x. [DOI] [PubMed] [Google Scholar]
- Knopf C.W. Evolution of viral DNA-dependent DNA polymerases. Virus Genes. 1998;16:47–58. doi: 10.1023/a:1007997609122. [DOI] [PubMed] [Google Scholar]
- Krakauer D.C., Jansen V.A.A., Jansen V.A.A. Red Queen dynamics of protein translation. J. Theor. Biol. 2002;218:97–109. doi: 10.1006/jtbi.2002.3054. [DOI] [PubMed] [Google Scholar]
- Kropinski A.M., Sibbald M.J., Sibbald M.J. Transfer RNA genes and their significance to codon usage in the Pseudomonas aeurginosa lambdoid bacteriophage D3. Can. J. Microbiol. 1999;45:791–796. [PubMed] [Google Scholar]
- Kunisawa T. Synonymous codon preferences in bacteriophage T4: A distinctive use of transfer RNAs from T4 and from its host Escherichia coli. J. Theor. Biol. 1992;159:287–298. doi: 10.1016/s0022-5193(05)80725-8. [DOI] [PubMed] [Google Scholar]
- Kunisawa T., Kanaya S., Kutter E., Kanaya S., Kutter E., Kutter E. Comparison of synonymous codon distribution patterns of bacteriophage and host genomes. DNA Res. 1998;5:319–326. doi: 10.1093/dnares/5.6.319. [DOI] [PubMed] [Google Scholar]
- Kurland C.G. Codon bias and gene expression. FEBS Lett. 1991;285:165–169. doi: 10.1016/0014-5793(91)80797-7. [DOI] [PubMed] [Google Scholar]
- Lawrence J.G., Hendrix R.W., Casjens S., Hendrix R.W., Casjens S., Casjens S. Where are the pseudogenes in bacterial genomes? Trends Microbiol. 2001;9:535–540. doi: 10.1016/s0966-842x(01)02198-9. [DOI] [PubMed] [Google Scholar]
- Lowe T., Eddy S., Eddy S. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E.S., Kutter E., Mosig G., Arisaka F., Kunisawa T., Ruger W., Kutter E., Mosig G., Arisaka F., Kunisawa T., Ruger W., Mosig G., Arisaka F., Kunisawa T., Ruger W., Arisaka F., Kunisawa T., Ruger W., Kunisawa T., Ruger W., Ruger W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A., Ochman H., Moran N.A., Ochman H., Moran N.A., Moran N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Muto A., Osawa S., Osawa S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc. Natl. Acad. Sci. 1987;84:166–169. doi: 10.1073/pnas.84.1.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J.A. Accounting for background nucleotide composition when measuring codon usage bias. Mol. Biol. Evol. 2002;19:1390–1394. doi: 10.1093/oxfordjournals.molbev.a004201. [DOI] [PubMed] [Google Scholar]
- Ohnishi M., Tanaka C., Kuhara S., Ishii K., Hattori M., Kurokawa K., Yasunaga T., Makino K., Shinagawa H., Murata T., Tanaka C., Kuhara S., Ishii K., Hattori M., Kurokawa K., Yasunaga T., Makino K., Shinagawa H., Murata T., Kuhara S., Ishii K., Hattori M., Kurokawa K., Yasunaga T., Makino K., Shinagawa H., Murata T., Ishii K., Hattori M., Kurokawa K., Yasunaga T., Makino K., Shinagawa H., Murata T., Hattori M., Kurokawa K., Yasunaga T., Makino K., Shinagawa H., Murata T., Kurokawa K., Yasunaga T., Makino K., Shinagawa H., Murata T., Yasunaga T., Makino K., Shinagawa H., Murata T., Makino K., Shinagawa H., Murata T., Shinagawa H., Murata T., Murata T., et al. Chromosome of the enterohemorrhagic Escherichia coli O157:H7; comparative analysis with K-12 MG1655 revealed the acquisition of a large amount of foreign DNAs. DNA Res. 1999;6:361–368. doi: 10.1093/dnares/6.6.361. [DOI] [PubMed] [Google Scholar]
- Raoult D., Audic S., Robert C., Abergel C., Renesto P., Ogata H., La Scola B., Suzan M., Claverie J.M., Audic S., Robert C., Abergel C., Renesto P., Ogata H., La Scola B., Suzan M., Claverie J.M., Robert C., Abergel C., Renesto P., Ogata H., La Scola B., Suzan M., Claverie J.M., Abergel C., Renesto P., Ogata H., La Scola B., Suzan M., Claverie J.M., Renesto P., Ogata H., La Scola B., Suzan M., Claverie J.M., Ogata H., La Scola B., Suzan M., Claverie J.M., La Scola B., Suzan M., Claverie J.M., Suzan M., Claverie J.M., Claverie J.M. The 1.2-megabase genome sequence of Mimivirus. Science. 2004;306:1344–1350. doi: 10.1126/science.1101485. [DOI] [PubMed] [Google Scholar]
- Rice P., Longden J., Bleasby A., Longden J., Bleasby A., Bleasby A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rocha E.P. Codon usage bias from tRNA’s point of view: Redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14:2279–2286. doi: 10.1101/gr.2896904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha E.P.C., Danchin A., Danchin A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002;18:291–294. doi: 10.1016/S0168-9525(02)02690-2. [DOI] [PubMed] [Google Scholar]
- Saporta G. Probabilités, Analyse des données et Statistique. Editions Technip; Paris, France: 1990. [Google Scholar]
- Sau K., Gupta S.K., Sau S., Ghosh T.C., Gupta S.K., Sau S., Ghosh T.C., Sau S., Ghosh T.C., Ghosh T.C. Synonymous codon usage bias in 16 Staphylococcus aureus phages: Implication in phage therapy. Virus Res. 2005;113:123–131. doi: 10.1016/j.virusres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Sharp P.M., Rogers M.S., McConnell D.J., Rogers M.S., McConnell D.J., McConnell D.J. Selection pressures on codon usage in the complete genome of bacteriophage T7. J. Mol. Evol. 1985;21:150–160. doi: 10.1007/BF02100089. [DOI] [PubMed] [Google Scholar]
- Sharp P.M., Bailes E., Grocock R.J., Peden J.F., Sockett R.E., Bailes E., Grocock R.J., Peden J.F., Sockett R.E., Grocock R.J., Peden J.F., Sockett R.E., Peden J.F., Sockett R.E., Sockett R.E. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005;33:1141–1153. doi: 10.1093/nar/gki242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle C.A. Viruses in the sea. Nature. 2005;437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- Turner P.E., Chao L., Chao L. Prisoner’s dilemma in an RNA virus. Nature. 1999;398:441–443. doi: 10.1038/18913. [DOI] [PubMed] [Google Scholar]
- Waldor M.K., Mekalanos J.J., Mekalanos J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- Weinbauer M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004;28:127–181. doi: 10.1016/j.femsre.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Weiss S.B., Hsu W.T., Foft J.W., Scherberg N.H., Hsu W.T., Foft J.W., Scherberg N.H., Foft J.W., Scherberg N.H., Scherberg N.H. Transfer RNA coded by the T4 bacteriophage genome. Proc. Natl. Acad. Sci. 1968;61:114–121. doi: 10.1073/pnas.61.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J.H. Function of the bacteriophage T4 transfer RNA’s. J. Mol. Biol. 1973;74:753–754. doi: 10.1016/0022-2836(73)90065-x. [DOI] [PubMed] [Google Scholar]